Objective ·To analyze the clinical and genetic characteristics of Chinese pediatric patients with Barth syndrome (BTHS) and provide data to support the prevention and treatment of BTHS. Methods ·Eighteen pediatric patients diagnosed with BTHS at Shanghai Children′s Medical Center, Shanghai Jiao Tong University School of Medicine, from January 2010 to November 2023, were included. Clinical data (age, birth weight, family history, electrocardiogram, echocardiogram, urine tandem mass spectrometry, complete blood count, blood biochemistry, and genetic test results) were collected to analyze the clinical characteristics, genetic findings, and prognoses of the patients. Results ·The study included 18 male patients with BTHS (including 2 monozygotic twins), consisting of one Yi ethnic and 17 Han Chinese patients. The median age at diagnosis was 3.0 (1.0, 5.6) months. Fifteen patients experienced decreased cardiac function at disease onset, with a left ventricular ejection fraction (LVEF) below 50%. Dilated cardiomyopathy (DCM) was observed in 15 patients, left ventricular non-compaction (LVNC) in 12 patients, and myocardial hypertrophy in 9 patients. During the diagnosis and follow-up, QTc interval prolongation occurred in 9 patients, ventricular arrhythmias in 2 patients, neutropenia in 9 patients, and monocytosis in 10 patients. Urine tandem mass spectrometry revealed 3-methylglutaconic aciduria (3-MGCA) in 8 of 13 tested patients. Fifteen types of TAZ gene mutation were identified in the 18 patients, including 5 novel mutations. Genetic testing of the parents of 16 patients indicated maternal inheritance in 15 cases. The median follow-up period was 8.5 (2.6, 29.3) months, during which 12 patients died. The median age at death was 7.5 (6.0, 12.8) months. Causes of death included heart failure (7 cases, with 4 concurrent infections), sudden death (3 cases), ventricular fibrillation (1 case), and accidental death (1 case). Conclusion ·BTHS is a rare genetic disorder with multisystem involvement. Its primary clinical manifestations include cardiomyopathy and neutropenia. The condition typically presents early in life, with severe progression and poor prognosis. Prompt recognition, accurate diagnosis, and early intervention are essential for managing this disease.
ZHAN Tianliu, YAN Zihang, WU Jinjin, CHEN Hao, CHEN Lijun, CHEN Yiwei, FU Lijun. Analysis of clinical and genetic characteristics of 18 pediatric patients with Barth syndrome. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2024, 44(11): 1406-1413 doi:10.3969/j.issn.1674-8115.2024.11.007
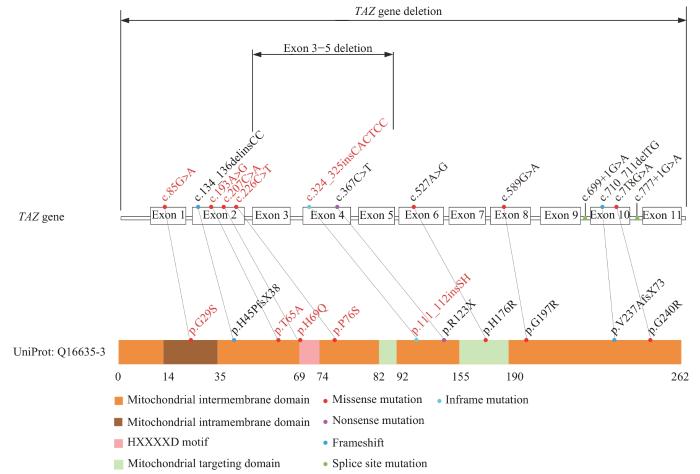
Barth综合征(Barth syndrome,BTHS)是一种由TAZ(G4.5)基因突变导致的X染色体隐性遗传的罕见病,由Peter G. Barth在1983年首次发现并报道[1],发病率为0.1/10万男性婴儿[2]。该病的患儿常在婴儿期发病,其临床特征以心肌病、骨骼肌病、中性粒细胞缺乏症和3-甲基戊烯二酸尿症(3-methylglutaconic aciduria,3-MGCA)多见。目前对BTHS的临床研究多基于国外高加索人种的数据[3],我国仅见散在的病例报道。本文回顾性分析18例BTHS患儿的临床特征和遗传学资料,以期为BTHS患儿的防治提供依据。
采集患儿的外周血,提取白细胞基因组DNA,进行医学外显子或全外显子高通量测序,利用Agilent SureSelect法进行外显子捕获,采用Illumina测序系统进行高通量测序,测序数据经NextGENe®软件分析后,用Ingenuity在线软件系统进行变异过筛及解释,候选变异经Sanger测序对家系成员进行验证,NM_000116.4作为TAZ基因编码区的参考序列。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)评级指南,对变异基因位点进行致病性分析。
The study was designed by ZHAN Tianliu, and FU Lijun. The manuscript was drafted and revised by ZHAN Tianliu, YAN Zihang and FU Lijun. The data were analyzed by ZHAN Tianliu and YAN Zihang. Data collection, case follow-up and study implementation were completed by CHEN Yiwei, WU Jinjin, CHEN Hao and CHEN Lijun. All the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突
COMPETING INTERESTS
All authors disclose no relevant conflict of interests.
BARTH P G, SCHOLTE H R, BERDEN J A, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes[J]. J Neurol Sci, 1983, 62(1/2/3): 327-355.
MILLER P C, REN M D, SCHLAME M, et al. A Bayesian analysis to determine the prevalence of Barth syndrome in the pediatric population[J]. J Pediatr, 2020, 217: 139-144.
ROBERTS A E, NIXON C, STEWARD C G, et al. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study[J]. Am J Med Genet A, 2012, 158A(11): 2726-2732.
LIPSHULTZ S E, LAW Y M, ASANTE-KORANG A, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association[J]. Circulation, 2019, 140(1): e9-e68.
JENNI R, OECHSLIN E, SCHNEIDER J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy[J]. Heart, 2001, 86(6): 666-671.
STEWARD C G, GROVES S J, TAYLOR C T, et al. Neutropenia in Barth syndrome: characteristics, risks, and management[J]. Curr Opin Hematol, 2019, 26(1): 6-15.
KANG S L, FORSEY J, DUDLEY D, et al. Clinical characteristics and outcomes of cardiomyopathy in Barth syndrome: the UK experience[J]. Pediatr Cardiol, 2016, 37(1): 167-176.
GARLID A O, SCHAFFER C T, KIM J, et al. TAZ encodes tafazzin, a transacylase essential for cardiolipin formation and central to the etiology of Barth syndrome[J]. Gene, 2020, 726: 144148.
D'ADAMO P, FASSONE L, GEDEON A, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies[J]. Am J Hum Genet, 1997, 61(4): 862-867.
CANTLAY A M, SHOKROLLAHI K, ALLEN J T, et al. Genetic analysis of the G4.5 gene in families with suspected Barth syndrome[J]. J Pediatr, 1999, 135(3): 311-315.
RIGAUD C, LEBRE A S, TOURAINE R, et al. Natural history of Barth syndrome: a national cohort study of 22 patients[J]. Orphanet J Rare Dis, 2013, 8: 70.
HIRONO K, HATA Y, NAKAZAWA M, et al. Clinical and echocardiographic impact of tafazzin variants on dilated cardiomyopathy phenotype in left ventricular non-compaction patients in early infancy[J]. Circ J, 2018, 82(10): 2609-2618.
THOMPSON W R, MANUEL R, ABBRUSCATO A, et al. Long-term efficacy and safety of elamipretide in patients with Barth syndrome: 168-week open-label extension results of TAZPOWER[J]. Genet Med, 2024, 26(7): 101138.
HORNBY B, THOMPSON W R, ALMUQBIL M, et al. Natural history comparison study to assess the efficacy of elamipretide in patients with Barth syndrome[J]. Orphanet J Rare Dis, 2022, 17(1): 336.
SCHAFER C, MOORE V, DASGUPTA N, et al. The effects of PPAR stimulation on cardiac metabolic pathways in Barth syndrome mice[J]. Front Pharmacol, 2018, 9: 318.
HUANG Y, POWERS C, MOORE V, et al. The PPAR pan-agonist bezafibrate ameliorates cardiomyopathy in a mouse model of Barth syndrome[J]. Orphanet J Rare Dis, 2017, 12(1): 49.
DABNER L, PIELES G E, STEWARD C G, et al. Treatment of Barth syndrome by cardiolipin manipulation (CARDIOMAN) with bezafibrate: protocol for a randomized placebo-controlled pilot trial conducted in the nationally commissioned Barth syndrome service[J]. JMIR Res Protoc, 2021, 10(5): e22533.
ZEGALLAI H M, HATCH G M. Barth syndrome: cardiolipin, cellular pathophysiology, management, and novel therapeutic targets[J]. Mol Cell Biochem, 2021, 476(3): 1605-1629.
LI Y, GODOWN J, TAYLOR C L, et al. Favorable outcomes after heart transplantation in Barth syndrome[J]. J Heart Lung Transplant, 2021, 40(10): 1191-1198.



{kind=link}
{kind=link}