Research progress and considerations for thalassemia gene therapy
GAO Xinjie,, LIU Yan,, WANG Dawei,
Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine (Shanghai), Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
Traditional treatment modalities for thalassemia include regular blood transfusions and allogenic hematopoietic stem cell transplantation (allo-HSCT). In recent years, autologous transplantation of gene-modified hematopoietic stem cells has emerged as a new curative strategy for transfusion-dependent thalassemia (TDT),which has the potential to replace conventional treatments, and provide lifelong benefits for patients. There are two existing technical approaches for gene therapy of β-thalassemia: gene addition, which involves transducing exogenous β-globin genes into hematopoietic stem cells (HSCs), and gene editing, which utilizes CRISPR-Cas9 or other editing systems to re-activate the expression of γ-globin gene. This article summarizes the marketed products and research progress in clinical trials, aiming to analyze the respective advantages and limitations of these two approaches, and discusses the effectiveness and safety of current gene therapies for β-thalassemia, as well as the future directions for associated technologies, including ex vivo HSC expansion with maintenance of stemness and vector-mediated in vivo gene modification. In terms of clinical translational medicine, this article provides in-depth insights into promising solutions for contemporary challenges confronted in clinical trials, including process development challenges, clinical trial conduct, regulatory approval processes, commercialization and payment systems.
GAO Xinjie, LIU Yan, WANG Dawei. Research progress and considerations for thalassemia gene therapy. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2025, 45(5): 540-548 doi:10.3969/j.issn.1674-8115.2025.05.002
临床上通过体征、区域调查、家族史和血液学检查(如血红蛋白电泳)快速筛查并诊断地中海贫血。目前,定期输血和祛铁治疗是TDT患者的主要治疗手段,但仅能维持生存和缓解症状。TDT患者需每2~5周输血1次,维持血红蛋白水平在95~105 g/L[2],终身治疗花费400~700万元人民币[9]。allo-HSCT是如今国内唯一能够治愈TDT的手段,由于移植成本高、年龄限制、全相合配型困难、移植后移植物抗宿主病(graft versus host disease,GVHD)和长期依赖免疫抑制剂等局限性,仅不到10%的患者真正获益[10]。
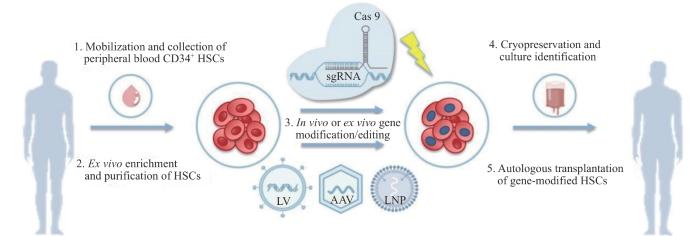
Fig 1
Schematic diagram of thalassemia gene therapy strategies
2.1 慢病毒介导的β-珠蛋白基因添加
首个基于LVV的β-地中海贫血基因治疗药物Zynteglo™(beti-cel)于2019年6月、2022年8月先后获得欧盟药物管理局(EMA)与美国食品和药物监督管理局(FDA)的上市许可,用于治疗≥12岁非β0/β0基因型的TDT患者。OTL-300采用骨内输注自体HSC疗法,Ⅰ/Ⅱ期试验中3例患儿摆脱输血依赖(transfusion independence,TI),并完成安全性和疗效的长期跟踪研究(NCT03275051)[11]。国内多家公司的LVV转导自体HSC产品已完成研究者发起的临床研究(investigator-initiated clinical trial,IIT),获批临床试验默示许可(investigational new drug,IND)并启动Ⅰ期临床试验,如HGI-001、KL003、BD211、GMCN508B(表1)。
Tab 1
表1
表1β-地中海贫血基因治疗的临床试验
Tab 1 Clinical trials of β-thalassemia gene therapy
Drug product
Sponsor
Status and
clinical trial identifier
Participant/n
Clinical trial result
Lentivirus-mediated β-globin gene addition
Zynteglo
Bluebird Bio
Phase Ⅰ/Ⅱ,
NCT01745120
22
Transfusion independence (TI) occurred in 12 patients with non-β0/β0 genotype and 3 patients with β0/β0 genotype or IVS1-110 homozygous mutation[12]
Phase Ⅲ,
NCT02906202
24
TI occurred in 20 patients with non-β0/β0 genotype while the average Hb level was 117 g/L, and the median level of HbAT87Q was 87 g/L at the 12th month after infusion[13]
Phase Ⅲ,
NCT03207009
19
One patient with β0/β0 genotype had TI for ≥ 12 months, while the Hb level of 3 patients with TI for ≥ 6 months was up to 105‒136 g/L (HbAT87Q accounting for 95‒126 g/L)[3]
OTL-300
Orchard Therapeutics
Phase Ⅰ/Ⅱ, NCT02453477
10
TI occurred in 3 pediatric patients, and red-cell transfusions were reduced in 4 patients[11]
HGI-001
Hemu Gene Co., Ltd.
IIT,
NCT05745532
10
The average Hb level of 5 patients was > 90 g/L during TI ≥ 12 months, with the median Hb level of 105 g/L at the most recent follow-up[14]
BD211
BDgene Co., Ltd.
IIT,
NCT05015920
10
Two patients with β0/β0 genotype had TI for 25.5 months in average, with red blood cell lifespan being extended to more than 42 d[15]
KL003
Kanglin Biotechnology
IIT,
ChiCTR2200055565
11
The average neutrophil and platelet engraftment times were both 14 d. TI occurred in all patients, with the longest duration lasting 18 months[16]
GMCN508B
Genmedicn Biopharma
IIT, NCT05762510
5
Two patients with TDT had TI, including a 10-year-old pediatric patient with β0/β+ genotype
Gene editing to re-activate the expression of HbF
ST-400
Sangamo Therapeutics
Phase Ⅰ/Ⅱ, NCT03432364
6
The low transduction efficiency of HSCs resulted in no long-term therapeutic effect[17]
CTX001
Vertex Pharmaceuticals
Phase Ⅲ,
NCT03655678
59
The median duration of TI among 48 patients was 22.5 months, with a total Hb level of 131 g/L and HbF level of 119 g/L in average[4]
EDIT-301
Editas Medicine
Phase Ⅰ/Ⅱ,
NCT05444894
9
TI occurred in 7 patients, including 6 with follow-up ≥ 6 months, whose total Hb and HbF levels were 121 g/L and 109 g/L, respectively[18]
ET-01
EdiGene Inc.
IIT, NCT04390971
3
One patient (β0/β+) had TI for>15 months, with a total Hb level of 110 g/L at the 18th month[19]
BRL-101
BRL Medicine
Phase Ⅰ,
NCT05577312
10
TI > 22 months occurred in all patients, including 5 with β0/β0 genotype. The highest HbF level reached 140 g/L, with HbF-cells accounting for 98%‒99%[20]
RM-001
Reforgene Medicine
Phase Ⅰ,
ChiCTR2300069244
12
Twelve patients in Phase Ⅰ and 7 patients in IIT study had TI for> 6 months (including 13 with β0/β0 genotype). Thirteen patients with ≥12 months of follow-up had an average HbF level of 117 g/L[21]
CS-101
CorrectSequence Therapeutics
IIT, NCT06291961
8
The first patient (β0/β+) had TI with HbF > 95 g/L at the 8th week
Casgevy™(exa-cel,CTX001)是全球首款CRISPR/Cas9基因编辑疗法,2024年1月获FDA批准上市,Ⅲ期临床试验(NCT04208529)中52例受试者正在接受15年的长期随访。与CTX001作用机制相似的还有基于锌指蛋白酶的ST-400,以及国内2款产品ET-01和BRL-101。破坏γ-珠蛋白启动子的BCL11A结合位点,也是重启HbF的策略,例如基于Cas9的RM-001、基于AsCas12a的EDIT-301、基于腺嘌呤碱基编辑器(adenine base editor,ABE)的BEAM-101,以及基于变形式碱基编辑器的CS-101(表1)。
碱基编辑器(base editor,BE)由dCas9或nCas9融合脱氨酶组成,有ABE(A:T to G:C)和CBE(C:G to T:A)2类,仅引入单碱基且无需DNA模板,不诱导DSB从而极大减少DDR,相较于HDR具有更高的编辑效率[40]。正序生物CS-101针对BCL11A结合位点进行C→T替换,已实现首例地中海贫血患者治愈。经脱氨酶改造的新一代BE,如hA3A-BE3、YEEBE4max[41]及ABE8[40]等编辑窗口改变且精确度提高。引导编辑器(prime editor,PE)能够以pegRNA为模板替换所有组合的碱基[41],比BE更灵活、更具特异性,已用于校正小鼠模型的IVS2-654突变,编辑效率达到14.29%[50]。
The review was designed by WANG Dawei, LIU Yan and GAO Xinjie. The manuscript was drafted by GAO Xinjie. WANG Dawei and LIU Yan participated in writing guidance and revision. All authors have read the last version of paper and consented to submission.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
All authors disclose no relevant conflict of interests.
Red Blood Cell Diseases (Anemia) Group, Chinese Society of Hematology, Chinese Medical Association. Chinese guideline for diagnosis and treatment of transfusion dependent β-thalassemia (2022)[J]. Chinese Journal of Hematology, 2022, 43(11): 889-896.
LAL A, LOCATELLI F, KWIATKOWSKI J L, et al. Northstar-3: interim results from a phase 3 study evaluating lentiglobin gene therapy in patients with transfusion-dependent β-thalassemia and either a β0 or IVS-I-110 mutation at both alleles of the HBB gene[J]. Blood, 2019, 134: 815.
Writing Group for Practice Guidelines for Diagnosis and Treatment of Genetic Diseases, Medical Genetics Branch of Chinese Medical Associatio. Clinical practice guidelines for β-thalassemia [J]. Chinese Journal of Medical Genetics, 2020, 37(3): 243-251.
ZHEN X M, MING J, ZHANG R Q, et al. Economic burden of adult patients with β-thalassaemia major in mainland China[J]. Orphanet J Rare Dis, 2023, 18(1): 252.
CHEN H, JIA Y Y, HUANG Y, et al. Progress and current status of gene therapy for thalassemia[J]. Journal of Guangxi Medical University, 2024, 41(1): 1-10.
THOMPSON A A, WALTERS M C, KWIATKOWSKI J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia[J]. N Engl J Med, 2018, 378(16): 1479-1493.
LOCATELLI F, THOMPSON A A, KWIATKOWSKI J L, et al. Betibeglogene autotemcel gene therapy for non-β0/β0 genotype β-thalassemia[J]. N Engl J Med, 2022, 386(5): 415-427.
HAN N. Interim results of gene therapy using optimized LentiHBBT87Q vector in five Chinese patients with transfusion dependent β-thalassemia[C]//EHA2024 Hybrid Congress. Madrid, Spain: EHA Library, 2024: 1517.
LI S Q, LING S K, WANG D W, et al. Modified lentiviral globin gene therapy for pediatric β0/β0 transfusion-dependent β-thalassemia: a single-center, single-arm pilot trial[J]. Cell Stem Cell, 2024, 31(7): 961-973.e8.
HUANG J Q, ZHANG Y M, LIANG L, et al. Gene therapy of transfusion-dependent β-thalassemia patients with quick engraftment of reinfused hematopoietic stem cells: an investigator-initiated trial of KL003[J]. Blood, 2023, 142: 4998.
WALTERS M C, SMITH A R, SCHILLER G J, et al. Updated results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent β thalassemia[J]. Blood, 2021, 138(Supplement 1): 3974.
FRANGOUL H, HANNA R, WALTERS M C, et al. Reni-cel, the first AsCas12a gene-edited cell therapy, shows promising preliminary results in key clinical outcomes in transfusion-dependent β- thalassemia patients treated in the EdiThaltrial[C]//EHA2024 Hybrid Congress. Madrid, Spain: EHA Library, 2024: 1476.
SHI J, FANG R G, GAO Z, et al. Preliminary safety and efficacy results of EDI001: an investigator initiated trial on CRISPR/Cas9-modified autologous CD34+ hematopoietic stem and progenitor cells for patients with transfusion dependent β-thalassemia[J]. Blood, 2022, 140(Supplement 1): 10652-10653.
ZHENG B, LIU R R, ZHANG X H, et al. Efficacy and safety of brl-101, CRISPR-Cas9-mediated gene editing of the BCL11A enhancer in transfusion-dependent β-thalassemia[J]. Blood, 2023, 142: 4995.
LIU R R, WANG L, XU H, et al. Safety and efficacy of RM-001 (autologous HBG1/2 promoter-modified CD34+ hematopoietic stem and progenitor cells) in patients with transfusion-dependent β-thalassemia[J]. Blood, 2023, 142: 4994.
MORGAN R A, UNTI M J, ALESHE B, et al. Improved titer and gene transfer by lentiviral vectors using novel, small β-globin locus control region elements[J]. Mol Ther, 2020, 28(1): 328-340.
GIOMMETTI A, PAPANIKOLAOU E. Advancements in hematopoietic stem cell gene therapy: a journey of progress for viral transduction[J]. Cells, 2024, 13(12): 1039.
ESRICK E B, LEHMANN L E, BIFFI A, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease[J]. N Engl J Med, 2021, 384(3): 205-215.
LIU B Y, BRENDEL C, VINJAMUR D S, et al. Development of a double shmiR lentivirus effectively targeting both BCL11A and ZNF410 for enhanced induction of fetal hemoglobin to treat β-hemoglobinopathies[J]. Mol Ther, 2022, 30(8): 2693-2708.
CAVAZZANA-CALVO M, PAYEN E, NEGRE O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia[J]. Nature, 2010, 467(7313): 318-322.
DUNCAN C N, BLEDSOE J R, GRZYWACZ B, et al. Hematologic cancer after gene therapy for cerebral adrenoleukodystrophy[J]. N Engl J Med, 2024, 391(14): 1287-1301.
DORMIANI K, MIR MOHAMMAD SADEGHI H, SADEGHI-ALIABADI H, et al. Long-term and efficient expression of human β-globin gene in a hematopoietic cell line using a new site-specific integrating non-viral system[J]. Gene Ther, 2015, 22(8): 663-674.
PFEIFER A, BRANDON E P, KOOTSTRA N, et al. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo[J]. Proc Natl Acad Sci USA, 2001, 98(20): 11450-11455.
BAHAL R, ALI MCNEER N, QUIJANO E, et al. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery[J]. Nat Commun, 2016, 7: 13304.
LAZARIS V M, SIMANTIRAKIS E, STAVROU E F, et al. Non-viral episomal vector mediates efficient gene transfer of the β-globin gene into K562 and human haematopoietic progenitor cells[J]. Genes (Basel), 2023, 14(9): 1774.
STAVROU E F, SIMANTIRAKIS E, VERRAS M, et al. Episomal vectors based on S/MAR and the β-globin replicator, encoding a synthetic transcriptional activator, mediate efficient γ-globin activation in haematopoietic cells[J]. Sci Rep, 2019, 9(1): 19765.
REES H A, MINELLA A C, BURNETT C A, et al. CRISPR-derived genome editing therapies: progress from bench to bedside[J]. Mol Ther, 2021, 29(11): 3125-3139.
ENACHE O M, RENDO V, ABDUSAMAD M, et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations[J]. Nat Genet, 2020, 52(7): 662-668.
FERRARI S, JACOB A, CESANA D, et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells[J]. Cell Stem Cell, 2022, 29(10): 1428-1444.e9.
LAMSFUS-CALLE A, DANIEL-MORENO A, UREÑA-BAILÉN G, et al. Universal gene correction approaches for β-hemoglobinopathies using CRISPR-Cas9 and adeno-associated virus serotype 6 donor templates[J]. CRISPR J, 2021, 4(2): 207-222.
NUALKAEW T, JEARAWIRIYAPAISARN N, HONGENG S, et al. Restoration of correct βIVS2-654-globin mRNA splicing and HbA production by engineered U7 snRNA in β-thalassaemia/HbE erythroid cells[J]. Sci Rep, 2019, 9(1): 7672.
KYLE CROMER M, CAMARENA J, MARTIN R M, et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells[J]. Nat Med, 2021, 27(4): 677-687.
PAVANI G, FABIANO A, LAURENT M, et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells[J]. Blood Adv, 2021, 5(5): 1137-1153.
LU D, XU Z L, PENG Z Y, et al. Induction of fetal hemoglobin by introducing natural hereditary persistence of fetal hemoglobin mutations in the γ-globin gene promoters for genome editing therapies for β-thalassemia[J]. Front Genet, 2022, 13: 881937.
SHYR D C, LOWSKY R, MILLER W, et al. One year follow-up on the first patient treated with nula-cel: an autologous CRISPR/Cas9 gene corrected CD34+ cell product to treat sickle cell disease[J]. Blood, 2023, 142: 5000.
ZHANG H K, SUN R L, FEI J, et al. Correction of β-thalassemia IVS-II-654 mutation in a mouse model using prime editing[J]. Int J Mol Sci, 2022, 23(11): 5948.
UCHIDA N, TISDALE J F, DONAHUE R E, et al. A single dose of CD117 antibody drug conjugate enables hematopoietic stem cell based gene therapy in nonhuman Primates[J]. Biol Blood Marrow Transplant, 2020, 26(3): S6.
NGUYEN G N, EVERETT J K, KAFLE S, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells[J]. Nat Biotechnol, 2021, 39(1): 47-55.
WANG H J, GEORGAKOPOULOU A, PSATHA N, et al. In vivo hematopoietic stem cell gene therapy ameliorates murine thalassemia intermedia[J]. J Clin Invest, 2019, 129(2): 598-615.
LI C, WANG H J, GEORGAKOPOULOU A, et al. In vivo HSC gene therapy using a bi-modular HDAd5/35++ vector cures sickle cell disease in a mouse model[J]. Mol Ther, 2021, 29(2): 822-837.
LI C, GEORGAKOPOULOU A, MISHRA A, et al. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice[J]. Blood Adv, 2021, 5(4): 1122-1135.
MEAKER G A, WILKINSON A C. Ex vivo hematopoietic stem cell expansion technologies: recent progress, applications, and open questions[J]. Exp Hematol, 2024, 130: 104136.
LI Y H, HE M, ZHANG W S, et al. Expansion of human megakaryocyte-biased hematopoietic stem cells by biomimetic Microniche[J]. Nat Commun, 2023, 14(1): 2207.
WANG H J, GEORGAKOPOULOU A, NIZAMIS E, et al. Auto-expansion of in vivo HDAd-transduced hematopoietic stem cells by constitutive expression of tHMGA2[J]. Mol Ther Methods Clin Dev, 2024, 32(3): 101319.
CORBACIOGLU S, FRANGOUL H, LOCATELLI F, et al. Defining curative endpoints for transfusion-dependent β-thalassemia in the era of gene therapy and gene editing[J]. Am J Hematol, 2024, 99(3): 422-429.
One patient with β0/β0 genotype had TI for ≥ 12 months, while the Hb level of 3 patients with TI for ≥ 6 months was up to 105‒136 g/L (HbAT87Q accounting for 95‒126 g/L)[3]
OTL-300
Orchard Therapeutics
Phase Ⅰ/Ⅱ, NCT02453477
10
TI occurred in 3 pediatric patients, and red-cell transfusions were reduced in 4 patients[11]
... 首个基于LVV的β-地中海贫血基因治疗药物Zynteglo™(beti-cel)于2019年6月、2022年8月先后获得欧盟药物管理局(EMA)与美国食品和药物监督管理局(FDA)的上市许可,用于治疗≥12岁非β0/β0基因型的TDT患者.OTL-300采用骨内输注自体HSC疗法,Ⅰ/Ⅱ期试验中3例患儿摆脱输血依赖(transfusion independence,TI),并完成安全性和疗效的长期跟踪研究(NCT03275051)[11].国内多家公司的LVV转导自体HSC产品已完成研究者发起的临床研究(investigator-initiated clinical trial,IIT),获批临床试验默示许可(investigational new drug,IND)并启动Ⅰ期临床试验,如HGI-001、KL003、BD211、GMCN508B(表1). ...
... NCT03207009
19
One patient with β0/β0 genotype had TI for ≥ 12 months, while the Hb level of 3 patients with TI for ≥ 6 months was up to 105‒136 g/L (HbAT87Q accounting for 95‒126 g/L)[3]
OTL-300
Orchard Therapeutics
Phase Ⅰ/Ⅱ, NCT02453477
10
TI occurred in 3 pediatric patients, and red-cell transfusions were reduced in 4 patients[11]
HGI-001
Hemu Gene Co., Ltd.
IIT, ...
1
... NCT01745120
22
Transfusion independence (TI) occurred in 12 patients with non-β0/β0 genotype and 3 patients with β0/β0 genotype or IVS1-110 homozygous mutation[12]
Phase Ⅲ, ...
1
... NCT02906202
24
TI occurred in 20 patients with non-β0/β0 genotype while the average Hb level was 117 g/L, and the median level of HbAT87Q was 87 g/L at the 12th month after infusion[13]
Phase Ⅲ, ...
1
... NCT05745532
10
The average Hb level of 5 patients was > 90 g/L during TI ≥ 12 months, with the median Hb level of 105 g/L at the most recent follow-up[14]
BD211
BDgene Co., Ltd.
IIT, ...
1
... NCT05015920
10
Two patients with β0/β0 genotype had TI for 25.5 months in average, with red blood cell lifespan being extended to more than 42 d[15]
KL003
Kanglin Biotechnology
IIT, ...
1
... ChiCTR2200055565
11
The average neutrophil and platelet engraftment times were both 14 d. TI occurred in all patients, with the longest duration lasting 18 months[16]
GMCN508B
Genmedicn Biopharma
IIT, NCT05762510
5
Two patients with TDT had TI, including a 10-year-old pediatric patient with β0/β+ genotype
Gene editing to re-activate the expression of HbF
ST-400
Sangamo Therapeutics
Phase Ⅰ/Ⅱ, NCT03432364
6
The low transduction efficiency of HSCs resulted in no long-term therapeutic effect[17]
CTX001
Vertex Pharmaceuticals
Phase Ⅲ, ...
1
... ChiCTR2200055565
11
The average neutrophil and platelet engraftment times were both 14 d. TI occurred in all patients, with the longest duration lasting 18 months[16]
GMCN508B
Genmedicn Biopharma
IIT, NCT05762510
5
Two patients with TDT had TI, including a 10-year-old pediatric patient with β0/β+ genotype
Gene editing to re-activate the expression of HbF
ST-400
Sangamo Therapeutics
Phase Ⅰ/Ⅱ, NCT03432364
6
The low transduction efficiency of HSCs resulted in no long-term therapeutic effect[17]
CTX001
Vertex Pharmaceuticals
Phase Ⅲ, ...
2
... NCT05444894
9
TI occurred in 7 patients, including 6 with follow-up ≥ 6 months, whose total Hb and HbF levels were 121 g/L and 109 g/L, respectively[18]
ET-01
EdiGene Inc.
IIT, NCT04390971
3
One patient (β0/β+) had TI for>15 months, with a total Hb level of 110 g/L at the 18th month[19]
TI occurred in 7 patients, including 6 with follow-up ≥ 6 months, whose total Hb and HbF levels were 121 g/L and 109 g/L, respectively[18]
ET-01
EdiGene Inc.
IIT, NCT04390971
3
One patient (β0/β+) had TI for>15 months, with a total Hb level of 110 g/L at the 18th month[19]
BRL-101
BRL Medicine
Phase Ⅰ, ...
1
... NCT05577312
10
TI > 22 months occurred in all patients, including 5 with β0/β0 genotype. The highest HbF level reached 140 g/L, with HbF-cells accounting for 98%‒99%[20]
RM-001
Reforgene Medicine
Phase Ⅰ, ...
2
... ChiCTR2300069244
12
Twelve patients in Phase Ⅰ and 7 patients in IIT study had TI for> 6 months (including 13 with β0/β0 genotype). Thirteen patients with ≥12 months of follow-up had an average HbF level of 117 g/L[21]
CS-101
CorrectSequence Therapeutics
IIT, NCT06291961
8
The first patient (β0/β+) had TI with HbF > 95 g/L at the 8th week
2.2 基因编辑重新激活<strong>HbF</strong>表达
Casgevy™(exa-cel,CTX001)是全球首款CRISPR/Cas9基因编辑疗法,2024年1月获FDA批准上市,Ⅲ期临床试验(NCT04208529)中52例受试者正在接受15年的长期随访.与CTX001作用机制相似的还有基于锌指蛋白酶的ST-400,以及国内2款产品ET-01和BRL-101.破坏γ-珠蛋白启动子的BCL11A结合位点,也是重启HbF的策略,例如基于Cas9的RM-001、基于AsCas12a的EDIT-301、基于腺嘌呤碱基编辑器(adenine base editor,ABE)的BEAM-101,以及基于变形式碱基编辑器的CS-101(表1). ...
... 碱基编辑器(base editor,BE)由dCas9或nCas9融合脱氨酶组成,有ABE(A:T to G:C)和CBE(C:G to T:A)2类,仅引入单碱基且无需DNA模板,不诱导DSB从而极大减少DDR,相较于HDR具有更高的编辑效率[40].正序生物CS-101针对BCL11A结合位点进行C→T替换,已实现首例地中海贫血患者治愈.经脱氨酶改造的新一代BE,如hA3A-BE3、YEEBE4max[41]及ABE8[40]等编辑窗口改变且精确度提高.引导编辑器(prime editor,PE)能够以pegRNA为模板替换所有组合的碱基[41],比BE更灵活、更具特异性,已用于校正小鼠模型的IVS2-654突变,编辑效率达到14.29%[50]. ...
... 碱基编辑器(base editor,BE)由dCas9或nCas9融合脱氨酶组成,有ABE(A:T to G:C)和CBE(C:G to T:A)2类,仅引入单碱基且无需DNA模板,不诱导DSB从而极大减少DDR,相较于HDR具有更高的编辑效率[40].正序生物CS-101针对BCL11A结合位点进行C→T替换,已实现首例地中海贫血患者治愈.经脱氨酶改造的新一代BE,如hA3A-BE3、YEEBE4max[41]及ABE8[40]等编辑窗口改变且精确度提高.引导编辑器(prime editor,PE)能够以pegRNA为模板替换所有组合的碱基[41],比BE更灵活、更具特异性,已用于校正小鼠模型的IVS2-654突变,编辑效率达到14.29%[50]. ...
... 碱基编辑器(base editor,BE)由dCas9或nCas9融合脱氨酶组成,有ABE(A:T to G:C)和CBE(C:G to T:A)2类,仅引入单碱基且无需DNA模板,不诱导DSB从而极大减少DDR,相较于HDR具有更高的编辑效率[40].正序生物CS-101针对BCL11A结合位点进行C→T替换,已实现首例地中海贫血患者治愈.经脱氨酶改造的新一代BE,如hA3A-BE3、YEEBE4max[41]及ABE8[40]等编辑窗口改变且精确度提高.引导编辑器(prime editor,PE)能够以pegRNA为模板替换所有组合的碱基[41],比BE更灵活、更具特异性,已用于校正小鼠模型的IVS2-654突变,编辑效率达到14.29%[50]. ...



{kind=link}
{kind=link}