Journal of Shanghai Jiao Tong University (Medical Science) ›› 2022, Vol. 42 ›› Issue (5): 609-616.doi: 10.3969/j.issn.1674-8115.2022.05.008
• Clinical research • Previous Articles Next Articles
ZHU Xiaowei1,2( ), ZHAN Feixia1, ZHANG Chao2, LIU Shihua2, ZHONG Ping2, CAO Li1,2, LUAN Xinghua1,2()
), ZHAN Feixia1, ZHANG Chao2, LIU Shihua2, ZHONG Ping2, CAO Li1,2, LUAN Xinghua1,2()
Received:2021-12-06
Accepted:2022-05-06
Online:2022-05-28
Published:2022-05-28
Contact:
LUAN Xinghua
E-mail:zhuxw@rjlab.cn;green_lxh@sina.com
Supported by:CLC Number:
ZHU Xiaowei, ZHAN Feixia, ZHANG Chao, LIU Shihua, ZHONG Ping, CAO Li, LUAN Xinghua. Analysis of genetic and clinical characteristics of PMP22-associated peripheral neuropathy[J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(5): 609-616.
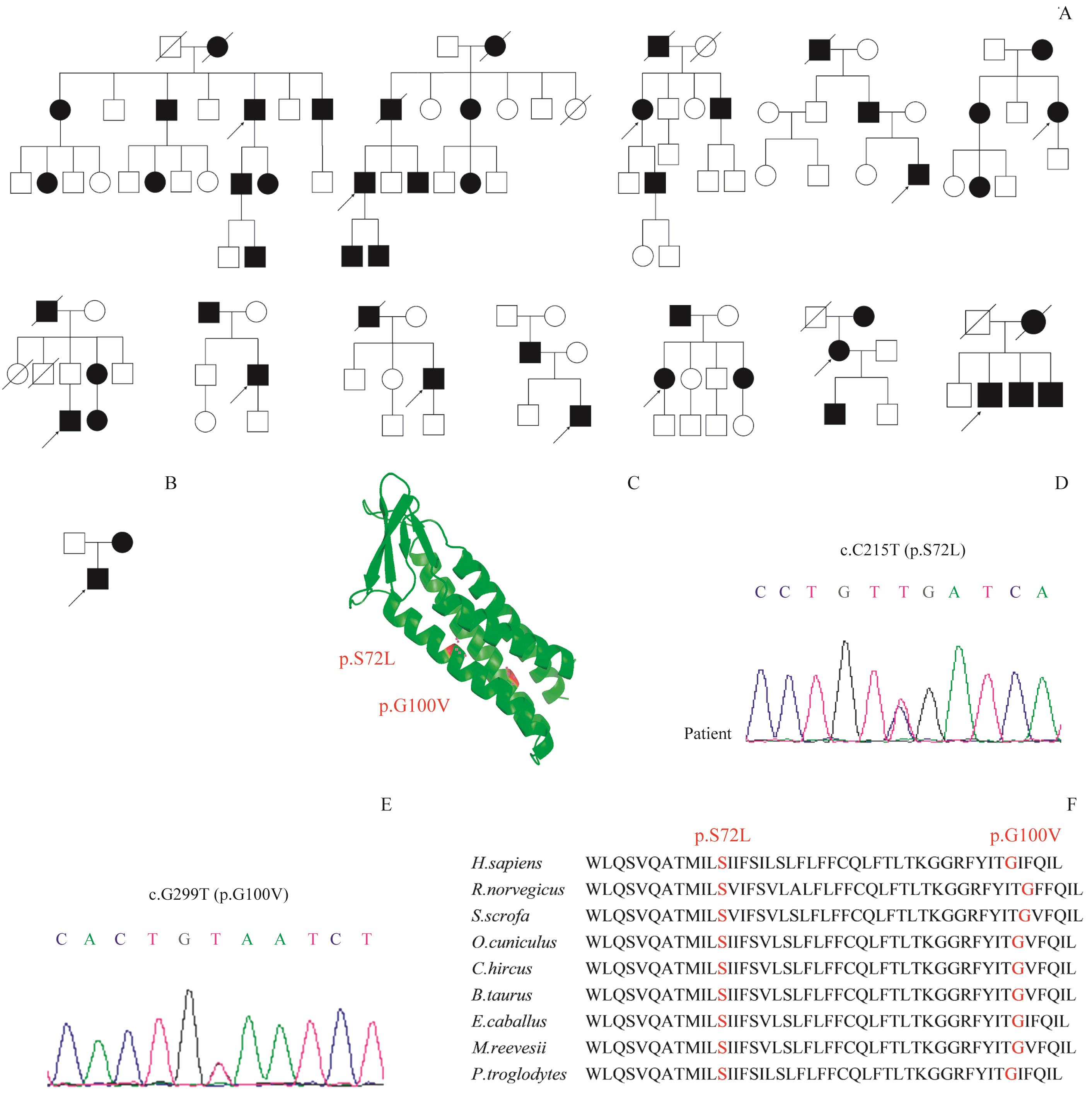
Fig 1 Family pedigrees and analysis of gene mutation
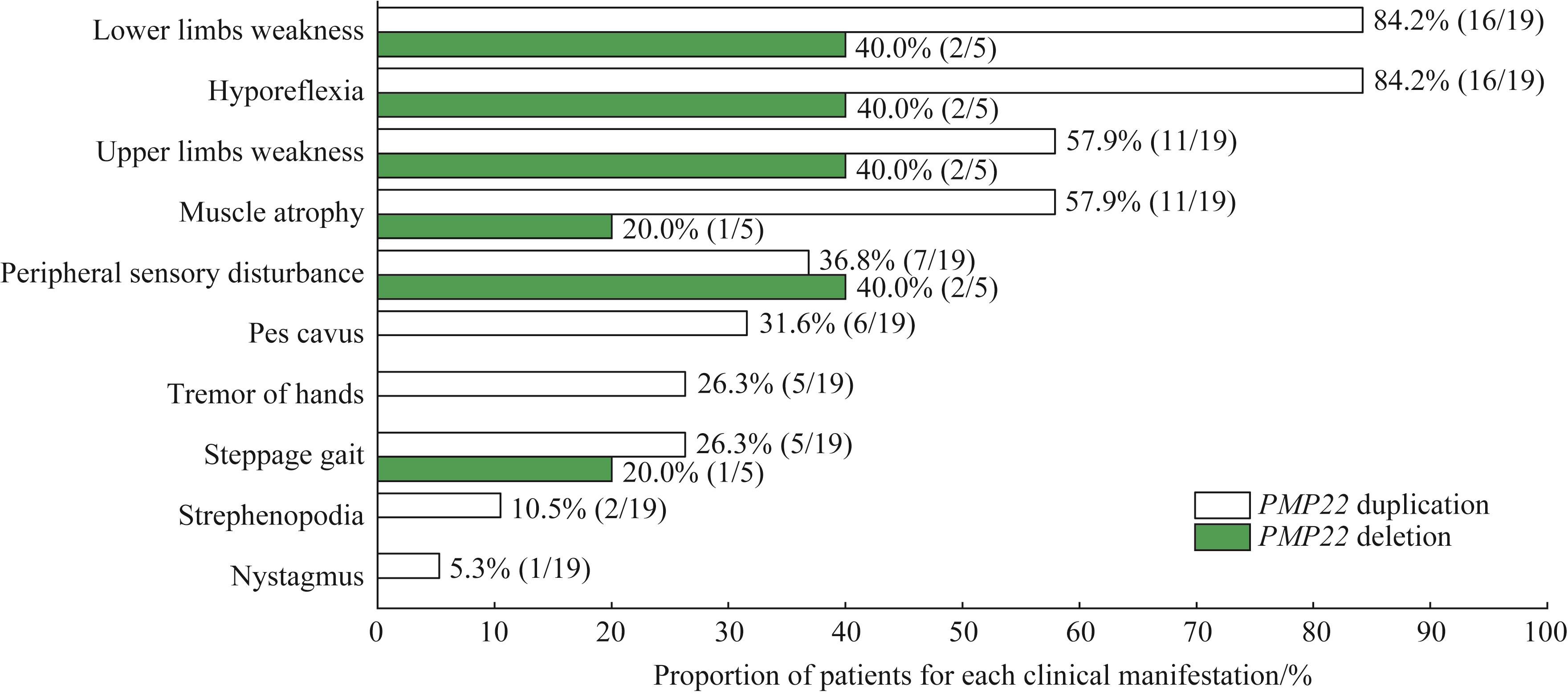
Fig 2 Clinical features of probands with PMP22 duplication and deletion mutations
| Index | PMP22 duplication (n=12) | P value① | PMP22 deletion (n=4) | P value① | P value② | ||
|---|---|---|---|---|---|---|---|
| Left | Right | Left | Right | ||||
| CMAP/mV | |||||||
| Median nerve (wrist-APB) | 3.401±1.559 | 3.434±1.886 | 0.972 | 6.100±2.823 | 7.275±1.227 | 0.553 | 0.008 |
| Median nerve (elbow-wrist) | 2.803±1.509 | 2.849±1.715 | 0.959 | 6.100±3.122 | 6.850±2.585 | 0.724 | 0.006 |
| Ulnar nerve (wrist-ADM) | 2.002±1.065 | 1.828±1.117 | 0.789 | 6.400±2.203 | 7.000±2.483 | 0.730 | 0.000 |
| Ulnar nerve (below elbow-wrist) | 1.487±1.087 | 1.243±0.951 | 0.688 | 6.075±2.095 | 6.475±2.294 | 0.805 | 0.000 |
| Tibial nerve (ankle-AH) | 16.375±1.576 | 16.775±3.163 | 0.661 | 39.033±0.850 | 37.533±2.774 | 0.421 | 0.000 |
| Common peroneal nerve (ankle-EDB) | 0.200±0.257 | 0.223±0.473 | 0.918 | 3.023±1.583 | 3.290±2.532 | 0.864 | 0.004 |
| MCV/(m·s-1) | |||||||
| Median nerve | 18.943±3.114 | 18.529±4.095 | 0.835 | 49.65±3.074 | 50.525±2.344 | 0.765 | 0.000 |
| Ulnar nerve | 17.833±2.420 | 18.767±3.266 | 0.586 | 50.900±4.809 | 50.675±8.207 | 0.964 | 0.000 |
| Tibial nerve | 0.315±0.574 | 0.343±0.346 | 0.920 | 7.533±2.937 | 8.633±2.754 | 0.661 | 0.001 |
| Common peroneal nerve | 14.143±3.629 | 17.100±4.964 | 0.227 | 36.775±2.136 | 35.425±4.751 | 0.623 | 0.000 |
| SCV/(m·s-1) | |||||||
| Median nerve | 12.843 (0.000, 310.200) | 8.210 (0.000, 33.600) | 0.548 | 41.575±2.990 | 41.475±2.666 | 0.962 | 0.000 |
| Ulnar nerve | 12.580 (0.000, 32.700) | 9.060 (0.000, 39.300) | 0.575 | 42.450±4.211 | 43.350±4.807 | 0.788 | 0.000 |
| Sural nerve | 9.691 (0.000, 34.600) | 7.792 (0.000, 27.600) | 0.717 | 40.850±8.009 | 41.575±7.374 | 0.898 | 0.000 |
Tab 1 Comparison of electrophysiological parameters between PMP22 duplication group and PMP22 deletion group
| Index | PMP22 duplication (n=12) | P value① | PMP22 deletion (n=4) | P value① | P value② | ||
|---|---|---|---|---|---|---|---|
| Left | Right | Left | Right | ||||
| CMAP/mV | |||||||
| Median nerve (wrist-APB) | 3.401±1.559 | 3.434±1.886 | 0.972 | 6.100±2.823 | 7.275±1.227 | 0.553 | 0.008 |
| Median nerve (elbow-wrist) | 2.803±1.509 | 2.849±1.715 | 0.959 | 6.100±3.122 | 6.850±2.585 | 0.724 | 0.006 |
| Ulnar nerve (wrist-ADM) | 2.002±1.065 | 1.828±1.117 | 0.789 | 6.400±2.203 | 7.000±2.483 | 0.730 | 0.000 |
| Ulnar nerve (below elbow-wrist) | 1.487±1.087 | 1.243±0.951 | 0.688 | 6.075±2.095 | 6.475±2.294 | 0.805 | 0.000 |
| Tibial nerve (ankle-AH) | 16.375±1.576 | 16.775±3.163 | 0.661 | 39.033±0.850 | 37.533±2.774 | 0.421 | 0.000 |
| Common peroneal nerve (ankle-EDB) | 0.200±0.257 | 0.223±0.473 | 0.918 | 3.023±1.583 | 3.290±2.532 | 0.864 | 0.004 |
| MCV/(m·s-1) | |||||||
| Median nerve | 18.943±3.114 | 18.529±4.095 | 0.835 | 49.65±3.074 | 50.525±2.344 | 0.765 | 0.000 |
| Ulnar nerve | 17.833±2.420 | 18.767±3.266 | 0.586 | 50.900±4.809 | 50.675±8.207 | 0.964 | 0.000 |
| Tibial nerve | 0.315±0.574 | 0.343±0.346 | 0.920 | 7.533±2.937 | 8.633±2.754 | 0.661 | 0.001 |
| Common peroneal nerve | 14.143±3.629 | 17.100±4.964 | 0.227 | 36.775±2.136 | 35.425±4.751 | 0.623 | 0.000 |
| SCV/(m·s-1) | |||||||
| Median nerve | 12.843 (0.000, 310.200) | 8.210 (0.000, 33.600) | 0.548 | 41.575±2.990 | 41.475±2.666 | 0.962 | 0.000 |
| Ulnar nerve | 12.580 (0.000, 32.700) | 9.060 (0.000, 39.300) | 0.575 | 42.450±4.211 | 43.350±4.807 | 0.788 | 0.000 |
| Sural nerve | 9.691 (0.000, 34.600) | 7.792 (0.000, 27.600) | 0.717 | 40.850±8.009 | 41.575±7.374 | 0.898 | 0.000 |
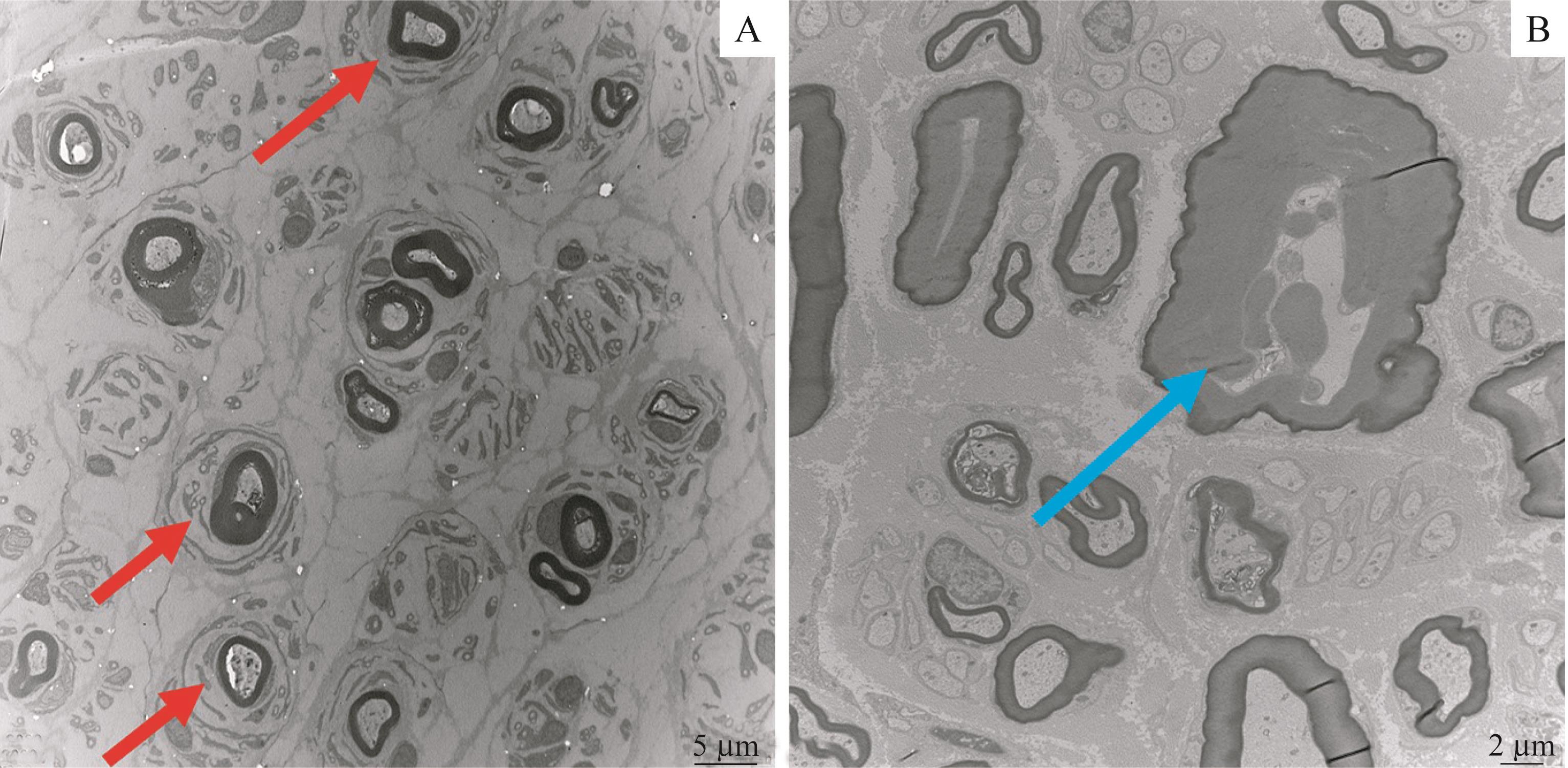
Fig 3 Sural nerve pathology of 1 CMT1A proband and 1 HNPPproband
| 1 | PANTERA H, MORAN J J, HUNG H A, et al. Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer[J]. Hum Mol Genet, 2018, 27(16): 2830-2839. |
| 2 | LIU X X, DUAN X H, ZHANG Y S, et al. Clinical and genetic diversity of PMP22 mutations in a large cohort of Chinese patients with Charcot-Marie-tooth disease[J]. Front Neurol, 2020, 11: 630. |
| 3 | 朱啸巍, 钟平, 栾兴华. PMP22相关性周围神经病的临床及遗传学特点[J]. 中国实用神经疾病杂志, 2021, 24(14): 1265-1270. |
| ZHU X W, ZHONG P, LUAN X H. The clinical and genetic characteristics of PMP22-associated peripheral neuropathy[J]. Chin J Pract Nerv Dis, 2021, 24(14): 1265-1270. | |
| 4 | GENTILE L, RUSSO M, FABRIZI G M, et al. Charcot-Marie-Tooth disease: experience from a large Italian tertiary neuromuscular center[J]. Neurol Sci, 2020, 41(5): 1239-1243. |
| 5 | WATILA M M, BALARABE S A. Molecular and clinical features of inherited neuropathies due to PMP22 duplication[J]. J Neurol Sci, 2015, 355(1/2): 18-24. |
| 6 | IVANOVIC V, BRANKOVIC M, BJELICA B, et al. Yield of the PMP22 deletion analysis in patients with compression neuropathies[J]. J Neurol, 2020, 267(12): 3617-3623. |
| 7 | D'ARRIGO S, TESSAROLLO V, TARONI F, et al. A case of severe early-onset neuropathy caused by a compound heterozygous deletion of the PMP22 gene: clinical and neurographic aspects[J]. Neuropediatrics, 2020, 51(3): 173-177. |
| 8 | MORINI A, MALAGUTI M C, MARANGONI S, et al. Neuropathic tremor in chronic inflammatory demyelinating polyneuropathy: the acquired equivalent of the roussy-levy syndrome[J]. Mov Disord Clin Pract, 2015, 3(2): 173-175. |
| 9 | 徐佳露, 章毅, 赵聪颖, 等. 13例早发型腓骨肌萎缩症的基因分型研究[J]. 中国当代儿科杂志, 2019, 21(7): 670-675. |
| XU J L, ZHANG Y, ZHAO C Y, et al. A genotyping study of 13 cases of early-onset Charcot-Marie-Tooth disease[J]. Chin J Contemp Pediat, 2019, 21(7): 670-675. | |
| 10 | ROA B B, DYCK P J, MARKS H G, et al. Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene[J]. Nat Genet, 1993, 5(3): 269-273. |
| 11 | MARQUES W Jr, THOMAS P K, SWEENEY M G, et al. Dejerine-Sottas neuropathy and PMP22 point mutations: a new base pair substitution and a possible “hot spot” on Ser72[J]. Ann Neurol, 1998, 43(5): 680-683. |
| 12 | RUSSO M, LAURÁ M, POLKE J M, et al. Variable phenotypes are associated with PMP22 missense mutations[J]. Neuromuscul Disord, 2011, 21(2): 106-114. |
| 13 | RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424. |
| 14 | WU R, LV H, ZHANG W, et al. Clinical and pathological variation of Charcot-Marie-tooth 1A in a large Chinese cohort[J]. Biomed Res Int, 2017, 2017: 6481367. |
| 15 | LOUSA M, VÁZQUEZ-HUARTE-MENDICOA C, GUTIÉRREZ A J, et al. Genetic epidemiology, demographic, and clinical characteristics of Charcot-Marie-tooth disease in the island of Gran Canaria (Spain)[J]. J Peripher Nerv Syst, 2019, 24(1): 131-138. |
| 16 | BROŽKOVÁ D, MAZANEC R, RYCHLÝ Z, et al. Four novel point mutations in the PMP22 gene with phenotypes of HNPP and Dejerine-Sottas neuropathy[J]. Muscle Nerve, 2011, 44(5): 819-822. |
| 17 | AZEVEDO H, PUPE C, PEREIRA R, et al. Pain in Charcot-Marie-Tooth disease: an update[J]. Arq Neuropsiquiatr, 2018, 76(4): 273-276. |
| 18 | KAWARAI T, YAMAZAKI H, MIYAMOTO R, et al. PMP22-related disease: a novel splice site acceptor variant and intrafamilial phenotype variability[J]. Neuromuscul Disord, 2019, 29(6): 422-426. |
| 19 | MITTENDORF K F, MARINKO J T, HAMPTON C M, et al. Peripheral myelin protein 22 alters membrane architecture[J]. Sci Adv, 2017, 3(7): e1700220. |
| 20 | ZHOU Y, MILES J R, TAVORI H, et al. PMP22 regulates cholesterol trafficking and ABCA1-mediated cholesterol efflux[J]. J Neurosci, 2019, 39(27): 5404-5418. |
| 21 | SVAREN J, MORAN J J, WU X Y, et al. Schwann cell transcript biomarkers for hereditary neuropathy skin biopsies[J]. Ann Neurol, 2019, 85(6): 887-898. |
| 22 | PANTERA H, SHY M E, SVAREN J. Regulating PMP22 expression as a dosage sensitive neuropathy gene[J]. Brain Res, 2020, 1726: 146491. |
| 23 | LI J. Genetic factors for nerve susceptibility to injuries: lessons from PMP22 deficiency[J]. Neural Regen Res, 2014, 9(18): 1661-1664. |
| 24 | TYSON J, ELLIS D, FAIRBROTHER U, et al. Hereditary demyelinating neuropathy of infancy. A genetically complex syndrome[J]. Brain, 1997, 120 ( Pt 1): 47-63. |
| 25 | NELIS E, HAITES N, VAN BROECKHOVEN C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies[J]. Hum Mutat, 1999, 13(1): 11-28. |
| 26 | WU R, FU J, MENG L C, et al. Homozygous splice-site mutation c.78+5G>A in PMP22 causes congenital hypomyelinating neuropathy[J]. Neuropathology, 2019, 39(6): 441-446. |
| 27 | LIAO Y C, TSAI P C, LIN T S, et al. Clinical and molecular characterization of PMP22 point mutations in Taiwanese patients with inherited neuropathy[J]. Sci Rep, 2017, 7(1): 15363. |
| 28 | RAMCHANDREN S. Charcot-Marie-tooth disease and other genetic polyneuropathies[J]. Continuum (Minneap Minn), 2017, 23(5, Peripheral Nerve and Motor Neuron Disorders): 1360-1377. |
| 29 | NAGAPPA M, SHARMA S, GOVINDARAJ P, et al. PMP22 gene-associated neuropathies: phenotypic spectrum in a cohort from India[J]. J Mol Neurosci, 2020, 70(5): 778-789. |
| 30 | ABE A, NAKAMURA K, KATO M, et al. Compound heterozygous PMP22 deletion mutations causing severe Charcot-Marie-Tooth disease type 1[J]. J Hum Genet, 2010, 55(11): 771-773. |
| 31 | AGOSTONI E, GOBESSI S, BRANCOLINI C, et al. Identification and characterization of a new member of the gas3/PMP22 gene family in C. elegans[J]. Gene, 1999, 234(2): 267-274. |
| 32 | ABE K T, LINO A M M, HIRATA M T A, et al. A novel stop codon mutation in the PMP22 gene associated with a variable phenotype[J]. Neuromuscul Disord, 2004, 14(5): 313-320. |
| 33 | TRACY J A, DYCK P J, KLEIN C J, et al. Onion-bulb patterns predict acquired or inherited demyelinating polyneuropathy[J]. Muscle Nerve, 2019, 59(6): 665-670. |
| 34 | MOSS K R, BOPP T S, JOHNSON A E, et al. New evidence for secondary axonal degeneration in demyelinating neuropathies[J]. Neurosci Lett, 2021, 744: 135595. |
| 35 | ZHAO H T, DAMLE S, IKEDA-LEE K, et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models[J]. J Clin Invest, 2018, 128(1): 359-368. |
| [1] | ZHANG Mi, LIU Shujun, LI Fubin, ZHANG Yan. Construction and evaluation of IgG antibody Fc combinational mutation library with mammalian cell surface display system [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(4): 510-517. |
| [2] | LUO Simin, YEAP Lengsiew, HAO Qian. Establishment and application of an in vitro detection method for antibody gene point mutation, insertion and deletion [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(4): 518-527. |
| [3] | Yun-fang MA, Li-na PAN, Zhen LI, Bei-li GAO, Jia-an HU, Zhi-hong XU. Exploratory study on downregulation of PD-L1 in KRAS G12V-mutant non-small cell lung cancer cells by selumetinib [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(6): 741-748. |
| [4] | Sheng CHENG, Yi ZHAO, Yong-chen WANG, Ping HUANG. Meta-analysis of the effect of BRAF gene mutation on the prognosis of colorectal cancer patients with liver metastases after hepatectomy [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(6): 786-792. |
| [5] | Yu-jie XIE, Li-juan XIE, Tian-wen ZHU, Yi-wen WANG. Study on interleukin-10 receptor A gene mutations-induced neonatal very early onset inflammatory bowel disease in 2 infants [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(3): 409-412. |
| [6] | ZHU Zhi-xing1, 2, JI Wei3, GU Jian-lei1, 4, LÜ Hui1, 2, 4, TIAN Guo-li3. Analysis of four mutations and protein structure of glucose-6-phosphate dehydrogenase gene [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2020, 40(12): 1571-1578. |
| [7] | WANG Jing, JIN Li-hui, ZHANG Qi, SUN Kun, YU Yu. ARHGEF16 variants screening and mutation function analysis for children with total anomalous pulmonary venous connection [J]. , 2020, 40(1): 70-. |
| [8] | LIU Si-jie,LI Ting-ting,CHEN Sun,LI Fen,SUN Kun,XU Rang. Mutation analysis of CITED2gene in patients with situs inversus [J]. , 2019, 39(5): 500-. |
| [9] | YAN Tian-qi1, CHEN Li-wei2, ZHU Yong-mei2, LI Jian-feng2, DAI Yu-ting2, 3, CUI Shu-Ya2, JIANG Lu2, CHEN Bing2, HUANG Jin-yan2. Construction of a knowledge database of gene fusion and mutation in acute lymphoblastic leukemia [J]. , 2018, 38(9): 1027-. |
| [10] | YANG Ying, SHI Xiao-dong, DAI Yu-jun, WANG Yue-ying. Rapamycin inhibiting the progression of acute myeloid leukemia with DNMT3Amutation [J]. , 2018, 38(9): 1039-. |
| [11] | LI Qing-li, WEN Jun, MIN Xue-jie, ZHAO Li, ZHAO Xiao-ping. Effect of IDHgene mutation on acute myeloid leukemia [J]. , 2018, 38(8): 960-. |
| [12] | WANG Bing-hua1, XU Wu-hen2, TANG Xiao-jun2, LAN Xiao-ping2. Analysis of KRT9 gene mutation in Chinese Han population [J]. , 2018, 38(12): 1425-. |
| [13] | LIU Gai-ling1, 2, TAO Kun3, DU Ai-lian2, LIU Xiao-li4, LIANG Hui1. Clinical characteristics and genetic analysis of nevoid basal cell carcinoma syndrome with epilepsy [J]. , 2018, 38(11): 1322-. |
| [14] | CHEN Jun-jue, TIAN Lin-lu, WEI Yan, KANG Xiao-li. Clinical characteristics of X-linked infantile nystagmus in a Chinese family [J]. , 2018, 38(11): 1355-. |
| [15] | HONG Sha1, ZHAO Dong-ying1, XIE Li-juan1, CHANG Guo-ying2, LIU Xiao-qing3, ZHU Tian-wen1. Analysis of two Chinese Han families with Duchenne/Becker muscular dystrophy [J]. , 2018, 38(10): 1223-. |
| Viewed | ||||||||||||||||||||||||||||||||||||||||||||||||||
|
Full text 877
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
Abstract 958
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||