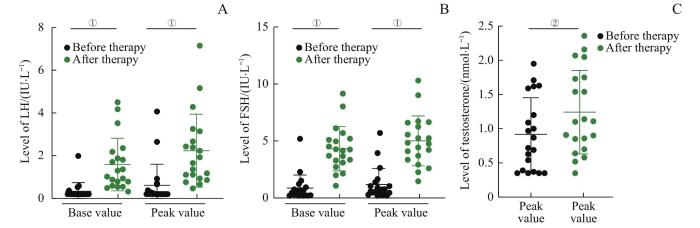
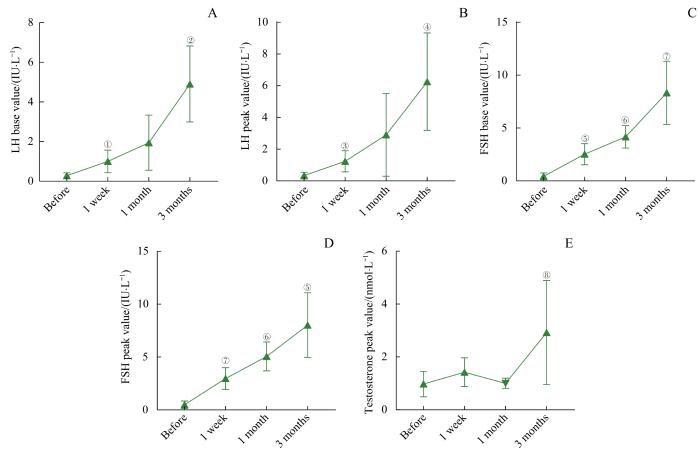
Objective ·To investigate the effect of short-term gonadotropin-releasing hormone (GnRH) pulse therapy on pituitary and testicular function in the adolescent male patients with congenital hypogonadotropic hypogonadism (CHH). Methods ·A retrospective study was conducted on 20 adolescent male patients with CHH who received GnRH pulse therapy from January 2016 to June 2021, and their clinical data were collected. They were treated with subcutaneous continuous pulsed administration of gonadorelin by the pump for 1 week (20 cases), of which 5 cases were treated for 3 months. The dose was 8‒10 μg per pulse, and the pulse interval was 90 min. The levels of luteinizing hormone (LH), follicle stimulating hormone (FSH) and testosterone were measured before GnRH pulse therapy at 1 week, 1 month and 3 months after treatment. After 3 months of treatment, the testicular volume was measured. All 20 patients with CHH underwent whole exome sequencing. Results ·The age of 20 CHH patients was 14.35 (14.08, 15.31) years old. The clinical manifestations were infantile testis (20/20) and micropenis (20/20), followed by obesity (12/20), dysosmia (9/20), insulin resistance (4/20), cryptorchidism (4/20), and short stature (3/20). The patients' height was 161.79 (154.90, 173.25) cm, body mass index was 23.80 (20.51, 27.46) kg/m2, and testicular volume was 0.91 (0.55, 1.25) mL. Inhibin B was 39.67 (11.29, 64.97) pg/mL; the base values of LH, FSH and testosterone before therapy were 0.20 (0.10, 0.30) IU/L, 0.87 (0.23, 0.89) IU/L, and 0.92 (0.38, 1.49) nmol/L, respectively. After 1 week of continuous GnRH pulse therapy, the base and peak values of LH and FSH and the peak value of testosterone in the 20 patients significantly increased (all P<0.05). In the 5 patients treated for 3 months, the base values and peak values of LH and FSH gradually increased with the prolongation of treatment time. After 3 months of treatment, the base values and peak values of LH and FSH, and the peak value of testosterone were significantly higher than those before treatment (all P<0.05), and the testicular volume was also significantly increased (P=0.004). Gene mutations were detected in only 14 of 20 patients, including fibroblast growth factor receptor 1 (FGFR1) mutations in 7 cases, anosmin 1 (ANOS1) mutations in 4 cases, prokineticin receptor 2 (PROKR2) mutations in 2 cases, and a prokineticin 2 (PROK2) mutation in 1 case. There was no significant difference of the effect of GnRH pulse therapy for 1 week on pituitary-testicular function between the patients with FGFR1 mutations and ANOS1 mutations. Conclusion ·The continuous GnRH pulse therapy for 1 week can make pituitary-testicular function respond in adolescent male CHH patients; the treatment for 3 months helps to induce the secondary sexual characteristics of puberty.
WANG Fei, GONG Yan, XU Liya, LIU Qingxu, LI Yan, GUO Sheng, LI Pin. Effect of short-term GnRH pulse therapy on pituitary-testicular function in adolescent male patients with congenital hypogonadotropic hypogonadism. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2023, 43(1): 36-43 doi:10.3969/j.issn.1674-8115.2023.01.005
全外显子组序列和拷贝数分析[8];平均覆盖深度>20×,覆盖广度为99.7%。使用ABI 3100测序仪对先证者的家庭成员及上海市儿童医院体检的100名健康儿童对照进行Sanger测序验证。应用DNAstar 5.0软件将结果与美国国家生物信息中心(National Center for Biotechnology Information,NCBI)参考序列进行比对,确定变异。新突变鉴定依据人类基因突变数据库(Human Gene Mutation Database,HGMD)、ClinVar数据库,以及外显子组整合数据库(Exome Aggregation Consortium,ExAC)。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类指南,评估变异分类。
Note: A. LH base value. B. LH peak value. C. FSH base value. D. FSH peak value. E. Testosterone level. ①P=0.023, ②P=0.005, ③P=0.014, ④P=0.012, ⑤P=0.006, ⑥P=0.022, ⑦P=0.004, ⑧P=0.045, compared with the level before GnRH pulse therapy.
Fig 2
Changes of LH, FSH and testosterone levels in the adolescent boys with CHH during the continuous GnRH pulse therapy for 3 months (n=5)
由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要。目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6]。CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型。卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16]。目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16]。本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变。其相关临床表型研究显示:① 垂体-睾丸功能障碍。曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差。FGFR1突变患者易出现隐睾[17]。本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异。② 嗅觉障碍。本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18]。③ 身高发育。本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符。由此可见,CHH基因型和临床表型之间并非简单的对应关系。本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱。
The study was designed by LI Pin and WANG Fei. WANG Fei, GONG Yan, LIU Qingxu, and LI Yan participated in the clinical data collection. LI Pin, GUO Sheng, and XU Liya participated in the treatment and follow-up. The manuscript was drafted and revised by LI Pin and WANG Fei. All the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
All authors disclose no relevant conflict of interests.
CANGIANO B, SWEE D S, QUINTON R, et al. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease[J]. Hum Genet, 2021, 140(1): 77-111.
SWEE D S, QUINTON R. Current concepts surrounding neonatal hormone therapy for boys with congenital hypogonadotropic hypogonadism[J]. Expert Rev Endocrinol Metab, 2022, 17(1): 47-61.
HAO M, NIE M, YU B Q, et al. Gonadotropin treatment for male partial congenital hypogonadotropic hypogonadism in Chinese patients[J]. Asian J Androl, 2020, 22(4): 390-395.
GONG C X, LIU Y, QIN M, et al. Pulsatile GnRH is superior to hCG in therapeutic efficacy in adolescent boys with hypogonadotropic hypogonadodism[J]. J Clin Endocrinol Metab, 2015, 100(7): 2793-2799.
SUN S Y, WANG W Q, JIANG Y R, et al. Treatment of idiopathic hypogonadotropic hypogonadism with pulse infusion of gonadorelin via micro pump[J]. Chinese Journal of Endocrinology and Metabolism, 2011, 27(8): 654-658.
GACH A, PINKIER I, SAŁACIŃSKA K, et al. Identification of gene variants in a cohort of hypogonadotropic hypogonadism: diagnostic utility of custom NGS panel and WES in unravelling genetic complexity of the disease[J]. Mol Cell Endocrinol, 2020, 517: 110968.
WANG Y, GONG C X, QIN M, et al. Clinical and genetic features of 64 young male paediatric patients with congenital hypogonadotropic hypogonadism[J]. Clin Endocrinol (Oxf), 2017, 87(6): 757-766.
MAIONE L, DWYER A A, FRANCOU B, et al. Genetics in endocrinology: genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing[J]. Eur J Endocrinol, 2018, 178(3): R55-R80.
AMATO L G L, MONTENEGRO L R, LERARIO A M, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism[J]. Eur J Endocrinol, 2019, 181(2): 103-119.
GACH A, PINKIER I, SZARRAS-CZAPNIK M, et al. Expanding the mutational spectrum of monogenic hypogonadotropic hypogonadism: novel mutations in ANOS1 and FGFR1 genes[J]. Reprod Biol Endocrinol, 2020, 18(1): 8.
LIU Q X, YIN X Q, LI P. Clinical, hormonal, and genetic characteristics of 25 Chinese patients with idiopathic hypogonadotropic hypogonadism[J]. BMC Endocr Disord, 2022, 22(1): 30.
FESTA A, UMANO G R, MIRAGLIA DEL GIUDICE E, et al. Genetic evaluation of patients with delayed puberty and congenital hypogonadotropic hypogonadism: is it worthy of consideration?[J]. Front Endocrinol (Lausanne), 2020, 11: 253.
KIM J H, SEO G H, KIM G H, et al. Targeted gene panel sequencing for molecular diagnosis of Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism[J]. Exp Clin Endocrinol Diabetes, 2019, 127(8): 538-544.
WANG Y, QIN M, FAN L J, et al. Correlation analysis of genotypes and phenotypes in Chinese male pediatric patients with congenital hypogonadotropic hypogonadism[J]. Front Endocrinol (Lausanne), 2022, 13: 846801.
LI S Y, ZHAO Y L, NIE M, et al. Clinical characteristics and spermatogenesis in patients with congenital hypogonadotropic hypogonadism caused by FGFR1 mutations[J]. Int J Endocrinol, 2020, 2020: 8873532.
NEOCLEOUS V, FANIS P, TOUMBA M, et al. GnRH deficient patients with congenital hypogonadotropic hypogonadism: novel genetic findings in ANOS1, RNF216, WDR11, FGFR1, CHD7, and POLR3A genes in a case series and review of the literature[J]. Front Endocrinol (Lausanne), 2020, 11: 626.
MOSBAH H, BOUVATTIER C, MAIONE L, et al. GnRH stimulation testing and serum inhibin B in males: insufficient specificity for discriminating between congenital hypogonadotropic hypogonadism from constitutional delay of growth and puberty[J]. Hum Reprod, 2020, 35(10): 2312-2322.
SEGAL T Y, MEHTA A, ANAZODO A, et al. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty[J]. J Clin Endocrinol Metab, 2009, 94(3): 780-785.
BINDER G, SCHWEIZER R, BLUMENSTOCK G, et al. Inhibin B plus LH vs GnRH agonist test for distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism in boys[J]. Clin Endocrinol (Oxf), 2015, 82(1): 100-105.
GAO Y T, DU Q, LIU L Y, et al. Serum inhibin B for differentiating between congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty: a systematic review and meta-analysis[J]. Endocrine, 2021, 72(3): 633-643.
VARIMO T, MIETTINEN P J, KÄNSÄKOSKI J, et al. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center[J]. Hum Reprod, 2017, 32(1): 147-153.
Division of Gonadal Disease, Chinese Society of Endocrinology. Expert consensus on the diagnosis and treatment of idiopathic hypogonadotropic hypogonadism[J]. Chinese Journal of Internal Medicine, 2015, 54(8): 739-744.
SHAH R, PATIL V, SARATHI V, et al. Prior testosterone replacement therapy may impact spermatogenic response to combined gonadotropin therapy in severe congenital hypogonadotropic hypogonadism[J]. Pituitary, 2021, 24(3): 326-333.
The Subspecialty Group of Endocrinologic, Hereditary and Metabolic Diseases, the Society of Pediatrics, Chinese Medical Association. Consensus statement on the diagnosis and endocrine treatment of children with disorder of sex development[J]. Chinese Journal of Pediatrics, 2019, 57(6): 410-418.
LIU Y, REN X Y, PENG Y G, et al. Efficacy and safety of human chorionic gonadotropin combined with human menopausal gonadotropin and a gonadotropin-releasing hormone pump for male adolescents with congenital hypogonadotropic hypogonadism[J]. Chin Med J (Engl), 2021, 134(10): 1152-1159.
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
... 全外显子组序列和拷贝数分析[8];平均覆盖深度>20×,覆盖广度为99.7%.使用ABI 3100测序仪对先证者的家庭成员及上海市儿童医院体检的100名健康儿童对照进行Sanger测序验证.应用DNAstar 5.0软件将结果与美国国家生物信息中心(National Center for Biotechnology Information,NCBI)参考序列进行比对,确定变异.新突变鉴定依据人类基因突变数据库(Human Gene Mutation Database,HGMD)、ClinVar数据库,以及外显子组整合数据库(Exome Aggregation Consortium,ExAC).根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类指南,评估变异分类. ...
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
2
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
... [11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
1
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
1
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
1
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
1
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
2
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...
1
... 由于部分CHH患者可能出现下丘脑-垂体-性腺轴功能的自行恢复,即可逆性CHH,且与体质性青春发育延迟(constitutional delay of growth and puberty,CDGP)之间存在重叠现象,因此遗传学分析尤为重要.目前已报道30余种CHH基因变异,但近50%遗传病因仍未明确[6].CHH根据有无嗅觉障碍分为嗅觉丧失型(即卡尔曼综合征)和嗅觉正常型.卡尔曼综合征占CHH的50%~60%,患病率约为1/4.8万[10-11],相关的基因有ANOS1、信号素3A(semaphorin 3A,SEMA3A)等[11-13];嗅觉正常型CHH相关的基因有kisspeptin 1蛋白(kisspeptin 1,KISS1)及其受体基因等[14-15];既可表现为嗅觉丧失型又可为正常型的CHH,相关基因有FGFR1、PROK2和PROKR2等[16].目前国内大样本研究报道CHH最常见的致病基因为FGFR1、PROKR2、CHD7(chromodomain helicase DNA binding protein 7)和ANOS1[16].本研究20例CHH患者中14例检测到基因突变,7例为FGFR1突变,4例为ANOS1突变,2例为PROKR2突变,1例为PROK2突变.其相关临床表型研究显示:① 垂体-睾丸功能障碍.曾有研究[9,12]报道ANOS1突变的卡尔曼综合征患者小阴茎、隐睾和幼稚型睾丸的发生率较高,治疗预后差.FGFR1突变患者易出现隐睾[17].本研究中7例FGFR1突变患者与4例ANOS1突变患者均存在小阴茎,各有1例隐睾;2种基因突变患者GnRH脉冲治疗1周后,垂体-睾酮功能的反应无明显差异.② 嗅觉障碍.本研究中9例CHH患者存在嗅觉障碍,分别携带ANOS1(2例)、FGFR1(2例)、PROK2(1例)和PROK2(1例)基因突变,另有3例未检测到基因突变,提示嗅觉障碍与基因型并不完全匹配,可能与可变外显率和差异表现度有关[1,18].③ 身高发育.本研究中FGFR1突变患者身高显著高于ANOS1突变患者,这与ANOS1突变临床表型较严重相符.由此可见,CHH基因型和临床表型之间并非简单的对应关系.本研究的对象因只纳入了接受GnRH泵治疗的患者,故不能完全体现CHH的突变基因谱. ...



{kind=link}
{kind=link}
{kind=link}
{kind=link}