Charcot-Marie-Tooth disease (CMT) is a group of hereditary motor and sensory neuropathy predominantly with peripheral neuropathy. It is characterized by progressive symmetric distal-predominant weakness, amyotrophy, sensory loss and reduced or absent deep tendon reflexes. CMT is usually divided into CMT1 type with demyelination and CMT2 type with axonal lesions according to electrophysiological and pathological characteristics. In addition to peripheral nervous system lesions, some CMT subtypes may also involve the central nervous system or other organs. The CMT patients with cerebellar system involvement also have cerebellar ataxia which can be seen as CMT1F type and CMT2E type caused by mutations in neurofilament light chain(NEFL) gene, CMT2Z with mutations in MORC family CW-type zinc finger 2 (MORC2) gene, CMT-6B with mutations in solute carrier family 25 member 46 (SLC25A46) gene, CMT2B2 with mutations in polynucleotide kinase 3′-phosphatase (PNKP) gene and so on. In recent years, CMT overlapping phenotypes have become a hot topic of research, among which CMT with cerebellar ataxia is a clinically and genetically heterogeneous group of disorders, and is prone to misdiagnosis clinically. This article reviews the clinical and genetic characteristics of CMT with cerebellar ataxia, aiming to provide reference for the earlier recognition and therapeutic strategies.
ZHU Xiaowei, ZHONG Ping, CAO Li, LUAN Xinghua. Clinical and genetic characteristics of Charcot-Marie-Tooth disease with cerebellar ataxia. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2023, 43(3): 350-357 doi:10.3969/j.issn.1674-8115.2023.03.011
目前为止,已有超过100个基因被确定为CMT致病基因。除周围神经组织之外,CMT也可能累及其他系统,如中枢神经系统、肌肉、骨骼和皮肤等,显示了CMT基因型-表现型的复杂性[2]。其中,小脑系统受累的CMT可见于神经丝蛋白轻链(neurofilament light chain,NEFL)突变所致的CMT1F型(OMIM#607734)和CMT2E型(OMIM#607684),MORC家族CW型锌指结构蛋白2(MORC family CW-type zinc finger 2,MORC2)突变所致的CMT2Z型(OMIM#616688),溶质载体家族25成员46(solute carrier family 25 member 46,SLC25A46)基因突变所致的伴视神经萎缩的CMT6B型(OMIM#616505),以及多核苷酸激酶3′-磷酸酶(polynucleotide kinase 3′-phosphatase,PNKP)基因突变所致的CMT2B2型(OMIM#605589)等。周围神经病变可能只是上述CMT表型的临床表现之一,本文对合并小脑性共济失调的CMT表型的临床及遗传学特点作一综述,旨在为CMT合并小脑性共济失调的诊治和鉴别提供参考。
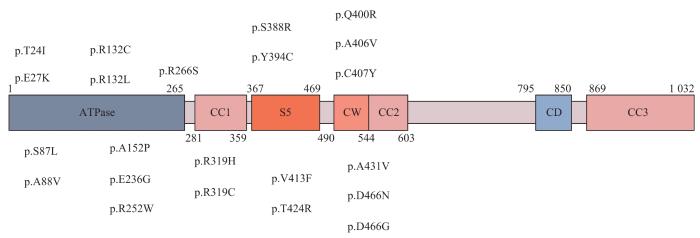
MORC2在中枢和外周神经的轴突和施万细胞中均有表达[24-25]。MORC2蛋白是ATP酶家族的成员,由N端GHKL(Gyrase B,Hsp90,histidine kinase and MutL)-ATP酶结构域、三链螺旋卷曲结构域、CW锌指结构域和靠近C端的双链卷曲结构域组成,大多数被发现的MORC2突变定位于GHKL-ATP酶结构域(图2)[26]。CMT2Z型发病机制相对更复杂,MORC2在过表达时可作为转录抑制因子,通过与其他基因相互作用导致多种表型的发生,也可通过与人类沉默中心(human silencing hub,HUSH)复合体相互作用,修饰染色质沉默表观遗传,在染色质重构、DNA修复和转录调控中发挥重要作用[23,26-29]。此外,细胞质中的MORC2可能参与脂质代谢和稳态维持[30]。研究[31]发现,MORC2可以下调精氨酸激酶结合蛋白2(arginine kinase-binding protein 2,ArgBP2)的表达。ArgBP2是一种肌动蛋白细胞骨架的衔接蛋白,定位于小脑突触后的浦肯野细胞[32]。在一项体外神经元模型(CMT2Z型患者成纤维细胞和啮齿动物感觉神经元)的研究中,研究者检测到神经递质受体和驱动蛋白基因的异常表达,MORC2突变的致病机制可能涉及编码轴突运输的蛋白质,与NEFL突变引起的CMT2E型类似[24]。
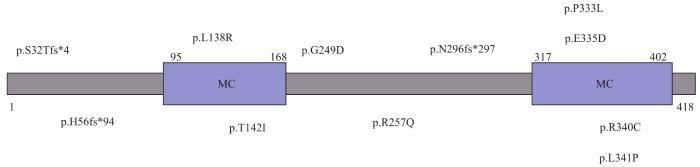
Fig 3
Structure diagram and mutation distribution of SLC25A46
3.2 临床表现
SLC25A46突变患者的发病年龄、临床特征和严重程度存在差异,主要表现为与小脑退行性变相关的视神经萎缩和轴索性神经病变[40,43]。患者在婴儿期或儿童期以视力障碍或平衡障碍起病,表现为视神经萎缩和进行性视力丧失,直到成年后才发展为周围神经病变,出现行走困难、远端肌肉萎缩、小脑性共济失调、眼球震颤、跨阈步态、语言困难、高弓足、脊柱侧凸畸形等表现[38,44]。此外,SLC25A46基因突变也可引起1E型脑桥小脑发育不全表型(pontocerebellar hypoplasia type 1E,PCH1E,OMIM#619303),与CMT6B型有重叠的临床特征,且发病时间越早,临床症状越严重[45]。
CMT2B2型神经电生理学表现与轴索型感觉运动神经病一致。上肢神经的MCV正常或轻微降低,下肢受累神经的MCV减慢、CMAP波幅减少、远端运动潜伏期(distal motor latencies,DML)增加,F波潜伏期相对保留。肌电图显示胫骨前肌群有明显异常,可观察到纤颤电位、多相电位增加、募集活动减少和宽时限、高波幅的运动单元动作电位,提示可能伴有活动性或慢性去神经支配[48,54]。
The manuscript was drafted and revised by ZHU Xiaowei. LUAN Xinghua reviewed and guided the revision of the paper. ZHONG Ping and CAO Li participated in the thesis conception and writing guidance. All the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
All authors disclose no relevant conflict of interests.
LAURÁ M, PIPIS M, ROSSOR A M, et al. Charcot-Marie-Tooth disease and related disorders: an evolving landscape[J]. Curr Opin Neurol, 2019, 32(5): 641-650.
ROSSOR A M, CARR A S, DEVINE H, et al. Peripheral neuropathy in complex inherited diseases: an approach to diagnosis[J]. J Neurol Neurosurg Psychiatry, 2017, 88(10): 846-863.
MERSIYANOVA I V, PEREPELOV A V, POLYAKOV A V, et al. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene[J]. Am J Hum Genet, 2000, 67(1): 37-46.
STONE E J, KOLB S J, BROWN A. A review and analysis of the clinical literature on Charcot-Marie-Tooth disease caused by mutations in neurofilament protein L[J]. Cytoskeleton (Hoboken), 2021, 78(3): 97-110.
ANTUNES L, FRASQUILHO S, OSTASZEWSKI M, et al. Similar α-synuclein staining in the colon mucosa in patients with Parkinson′s disease and controls[J]. Mov Disord, 2016, 31(10): 1567-1570.
ZHAI J B, LIN H, JULIEN J P, et al. Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1[J]. Hum Mol Genet, 2007, 16(24): 3103-3116.
MARTIN M, IYADURAI S J, GASSMAN A, et al. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport[J]. Mol Biol Cell, 1999, 10(11): 3717-3728.
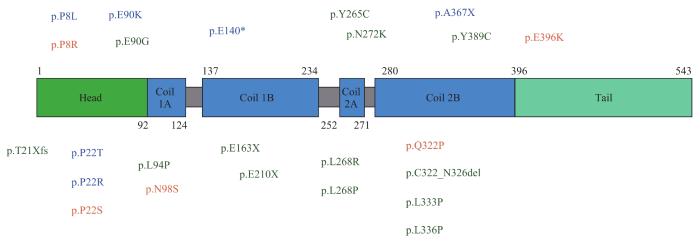
YUM S W, ZHANG J X, MO K T, et al. A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy[J]. Ann Neurol, 2009, 66(6): 759-770.
LERAT J, MAGDELAINE C, BEAUVAIS-DZUGAN H, et al. A novel pathogenic variant of NEFL responsible for deafness associated with peripheral neuropathy discovered through next-generation sequencing and review of the literature[J]. J Peripher Nerv Syst, 2019, 24(1): 139-144.
GEORGIOU D M, ZIDAR J, KOROSEC M, et al. A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family[J]. Neurogenetics, 2002, 4(2): 93-96.
FABRIZI G M, CAVALLARO T, ANGIARI C, et al. Giant axon and neurofilament accumulation in Charcot-Marie-Tooth disease type 2E[J]. Neurology, 2004, 62(8): 1429-1431.
KIM H J, KIM S B, KIM H S, et al. Phenotypic heterogeneity in patients with NEFL-related Charcot-Marie-Tooth disease[J]. Mol Genet Genomic Med, 2022, 10(2): e1870.
LIKAR T, HASANHODŽIĆ M, TERAN N, et al. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss[J]. PLoS One, 2018, 13(1): e0188578.
PISCIOTTA C, BAI Y H, BRENNAN K M, et al. Reduced neurofilament expression in cutaneous nerve fibers of patients with CMT2E[J]. Neurology, 2015, 85(3): 228-234.
AGRAWAL P B, JOSHI M, MARINAKIS N S, et al. Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings[J]. JAMA Neurol, 2014, 71(11): 1413-1420.
HORGA A, LAURÀ M, JAUNMUKTANE Z, et al. Genetic and clinical characteristics of NEFL-related Charcot-Marie-Tooth disease[J]. J Neurol Neurosurg Psychiatry, 2017, 88(7): 575-585.
DOPPLER K, KUNSTMANN E, KRÜGER S, et al. Painful Charcot-Marie-Tooth neuropathy type 2E/1F due to a novel NEFL mutation[J]. Muscle Nerve, 2017, 55(5): 752-755.
SMITH S M, JENKINSON M, JOHANSEN-BERG H, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data[J]. NeuroImage, 2006, 31(4): 1487-1505.
HWANG S, PARK C H, KIM R E, et al. Cerebellar white matter abnormalities in Charcot-Marie-Tooth Disease: a combined volumetry and diffusion tensor imaging analysis[J]. J Clin Med, 2021, 10(21): 4945.
ANDO M, OKAMOTO Y, YOSHIMURA A, et al. Clinical and mutational spectrum of Charcot-Marie-Tooth disease type 2Z caused by MORC2 variants in Japan[J]. Eur J Neurol, 2017, 24(10): 1274-1282.
GUILLEN SACOTO M J, TCHASOVNIKAROVA I A, TORTI E, et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism[J]. Am J Hum Genet, 2020, 107(2): 352-363.
WANG G L, WANG C Y, CAI X Z, et al. Identification and expression analysis of a novel CW-type zinc finger protein MORC2 in cancer cells[J]. Anat Rec (Hoboken), 2010, 293(6): 1002-1009.
TCHASOVNIKAROVA I A, TIMMS R T, DOUSE C H, et al. Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2[J]. Nat Genet, 2017, 49(7): 1035-1044.
LI D Q, NAIR S S, OHSHIRO K, et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response[J]. Cell Rep, 2012, 2(6): 1657-1669.
DOUSE C H, BLOOR S, LIU Y C, et al. Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms[J]. Nat Commun, 2018, 9(1): 651.
SHAO Y G, LI Y, ZHANG J, et al. Involvement of histone deacetylation in MORC2-mediated down-regulation of carbonic anhydrase Ⅸ[J]. Nucleic Acids Res, 2010, 38(9): 2813-2824.
CESTRA G, TOOMRE D, CHANG S, et al. The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton[J]. Proc Natl Acad Sci U S A, 2005, 102(5): 1731-1736.
ZANNI G, NARDELLA M, BARRESI S, et al. De novo p.T362R mutation in MORC2 causes early onset cerebellar ataxia, axonal polyneuropathy and nocturnal hypoventilation[J]. Brain, 2017, 140(6): e34.
HYUN Y S, HONG Y B, CHOI B O, et al. Clinico-genetics in Korean Charcot-Marie-Tooth disease type 2Z with MORC2 mutations[J]. Brain, 2016, 139(Pt 7): e40.
RAJU S, MEDARAMETLA S, BORAIAH N. Dystonia and hereditary motor sensory neuropathy 6B due to SLC25A46 gene mutations[J]. Mov Disord Clin Pract, 2021, 8(3): 480-482.
ABRAMS A J, HUFNAGEL R B, REBELO A, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder[J]. Nat Genet, 2015, 47(8): 926-932.
PERIVOLIDI V I, VIOLITZI F, IOANNIDOU E, et al. Proteomic identification of the SLC25A46 interactome in transgenic mice expressing SLC25A46-FLAG[J]. J Proteome Res, 2022, 21(2): 375-394.
WAN J J, STEFFEN J, YOURSHAW M, et al. Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia[J]. Brain, 2016, 139(11): 2877-2890.
GAO L, WANG M, LIAO L F, et al. A Slc25a46 mouse model simulating age-associated motor deficit, redox imbalance, and mitochondria dysfunction[J]. J Gerontol A Biol Sci Med Sci, 2021, 76(3): 440-447.
LI Z, PENG Y Y, HUFNAGEL R B, et al. Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice[J]. Hum Mol Genet, 2017, 26(19): 3776-3791.
NGUYEN M, BOESTEN I, HELLEBREKERS D M, et al. Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder[J]. Clin Genet, 2017, 91(1): 121-125.
ABRAMS A J, FONTANESI F, TAN N B L, et al. Insights into the genotype-phenotype correlation and molecular function of SLC25A46[J]. Hum Mutat, 2018, 39(12): 1995-2007.
BRAUNISCH M C, GALLWITZ H, ABICHT A, et al. Extension of the phenotype of biallelic loss-of-function mutations in SLC25A46 to the severe form of pontocerebellar hypoplasia type Ⅰ[J]. Clin Genet, 2018, 93(2): 255-265.
CHARLESWORTH G, BALINT B, MENCACCI N E, et al. SLC25A46 mutations underlie progressive myoclonic ataxia with optic atrophy and neuropathy[J]. Mov Disord, 2016, 31(8): 1249-1251.
LEAL A, BOGANTES-LEDEZMA S, EKICI A B, et al. The polynucleotide kinase 3′-phosphatase gene (PNKP) is involved in Charcot-Marie-Tooth disease (CMT2B2) previously related to MED25[J]. Neurogenetics, 2018, 19(4): 215-225.
LEAL A, MORERA B, DEL VALLEG, et al. A second locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 19q13.3[J]. Am J Hum Genet, 2001, 68(1): 269-274.
JILANI A, RAMOTAR D, SLACK C, et al. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage[J]. J Biol Chem, 1999, 274(34): 24176-24186.
BERNSTEIN N K, WILLIAMS R S, RAKOVSZKY M L, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase[J]. Mol Cell, 2005, 17(5): 657-670.
TSUKADA K, MATSUMOTO Y, SHIMADA M. Linker region is required for efficient nuclear localization of polynucleotide kinase phosphatase[J]. PLoS One, 2020, 15(9): e0239404.
PEDROSO J L, ROCHA C R, MACEDO-SOUZA L I, et al. Mutation in PNKP presenting initially as axonal Charcot-Marie-Tooth disease[J]. Neurol Genet, 2015, 1(4): e30.
BERGHOFF C, BERGHOFF M, LEAL A, et al. Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2B) that maps to chromosome 19q13.3[J]. Neuromuscul Disord, 2004, 14(5): 301-306.
DJORDJEVIC D, PINARD M, GAUTHIER M S, et al. De novo variants in POLR3B cause ataxia, spasticity, and demyelinating neuropathy[J]. Am J Hum Genet, 2022, 109(4): 759-763.
HYUN Y S, PARK H J, HEO S H, et al. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy[J]. Clin Genet, 2014, 86(6): 592-594.
CORTESE A, SIMONE R, SULLIVAN R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia[J]. Nat Genet, 2019, 51(4): 649-658.
LI Z Y, TIAN W T, SHEN J Y, et al. Atypical autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of two families[J]. Chinese Journal of Nervous and Mental Diseases, 2021, 47(8): 509-512.
... CMT2B2型神经电生理学表现与轴索型感觉运动神经病一致.上肢神经的MCV正常或轻微降低,下肢受累神经的MCV减慢、CMAP波幅减少、远端运动潜伏期(distal motor latencies,DML)增加,F波潜伏期相对保留.肌电图显示胫骨前肌群有明显异常,可观察到纤颤电位、多相电位增加、募集活动减少和宽时限、高波幅的运动单元动作电位,提示可能伴有活动性或慢性去神经支配[48,54]. ...
... CMT2B2型神经电生理学表现与轴索型感觉运动神经病一致.上肢神经的MCV正常或轻微降低,下肢受累神经的MCV减慢、CMAP波幅减少、远端运动潜伏期(distal motor latencies,DML)增加,F波潜伏期相对保留.肌电图显示胫骨前肌群有明显异常,可观察到纤颤电位、多相电位增加、募集活动减少和宽时限、高波幅的运动单元动作电位,提示可能伴有活动性或慢性去神经支配[48,54]. ...




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}