上海交通大学学报(医学版) ›› 2023, Vol. 43 ›› Issue (3): 350-357.doi: 10.3969/j.issn.1674-8115.2023.03.011
• 综述 • 上一篇
朱啸巍1,2( ), 钟平2, 曹立1,2, 栾兴华1,2()
), 钟平2, 曹立1,2, 栾兴华1,2()
收稿日期:2022-07-29
接受日期:2023-03-01
出版日期:2023-03-28
发布日期:2023-03-28
通讯作者:
栾兴华
E-mail:zhuxw@rjlab.cn;green_lxh@sina.com
作者简介:朱啸巍(1997—),女,硕士生;电子信箱:zhuxw@rjlab.cn。
基金资助:
ZHU Xiaowei1,2(), ZHONG Ping2, CAO Li1,2, LUAN Xinghua1,2()
Received:2022-07-29
Accepted:2023-03-01
Online:2023-03-28
Published:2023-03-28
Contact:
LUAN Xinghua
E-mail:zhuxw@rjlab.cn;green_lxh@sina.com
Supported by:摘要:
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)是一组以周围神经病变为主的遗传性运动感觉神经病。主要临床症状包括进行性对称性肢体远端无力、萎缩、感觉障碍和腱反射减退或消失。根据神经电生理表现和病理特点,CMT可分为以脱髓鞘为主的CMT1型和轴索病变为主的CMT2型。除了周围神经系统病变外,CMT部分表型可同时累及中枢神经系统或其他脏器;其中小脑系统受累的CMT患者同时合并小脑性共济失调,可见于神经丝蛋白轻链(neurofilament light chain,NEFL)基因突变所致的CMT1F型和CMT2E型,MORC家族CW型锌指结构蛋白2(MORC family CW-type zinc finger 2,MORC2)基因突变所致的CMT2Z型,溶质载体家族25成员46(solute carrier family 25 member 46,SLC25A46)基因突变所致的伴视神经萎缩的CMT6B型,以及多核苷酸激酶3′-磷酸酶(polynucleotide kinase 3′-phosphatase,PNKP)基因突变所致的CMT2B2型等。近年来,CMT重叠表型成为研究的热点,其中CMT合并小脑性共济失调具有高度临床异质性和遗传异质性,临床上易发生误诊。该文就合并小脑性共济失调的CMT表型的临床及遗传学特点进行综述,旨在为该类患者的早期诊断和治疗提供参考。
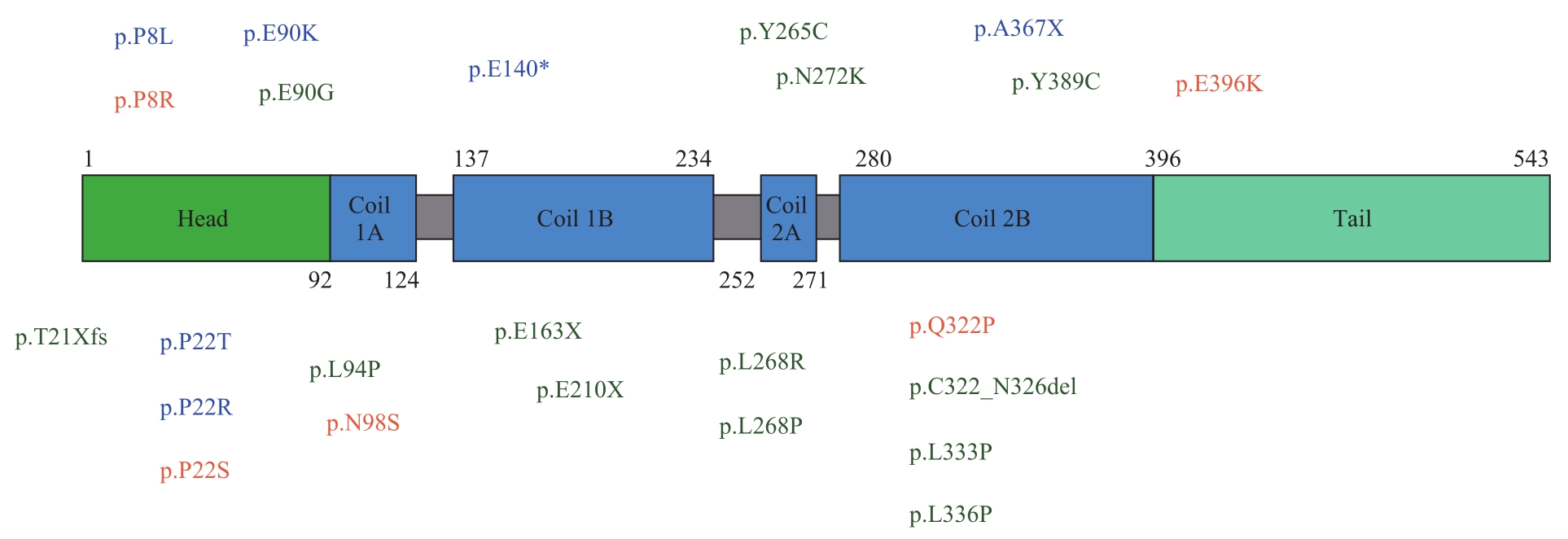
图1 NEFL的结构示意图及突变分布Note: CMT1F mutations are shown in blue; CMT2E mutations are shown in green and both in orange.
Fig 1 Structure diagram and mutation distribution of NEFL

图2 MORC2的结构示意图及突变分布Note: CC—coiled coil; S5—transducer S5-like domain; CW—zinc finger domain; CD—coil domain.
Fig 2 Structure diagram and mutation distribution of MORC2
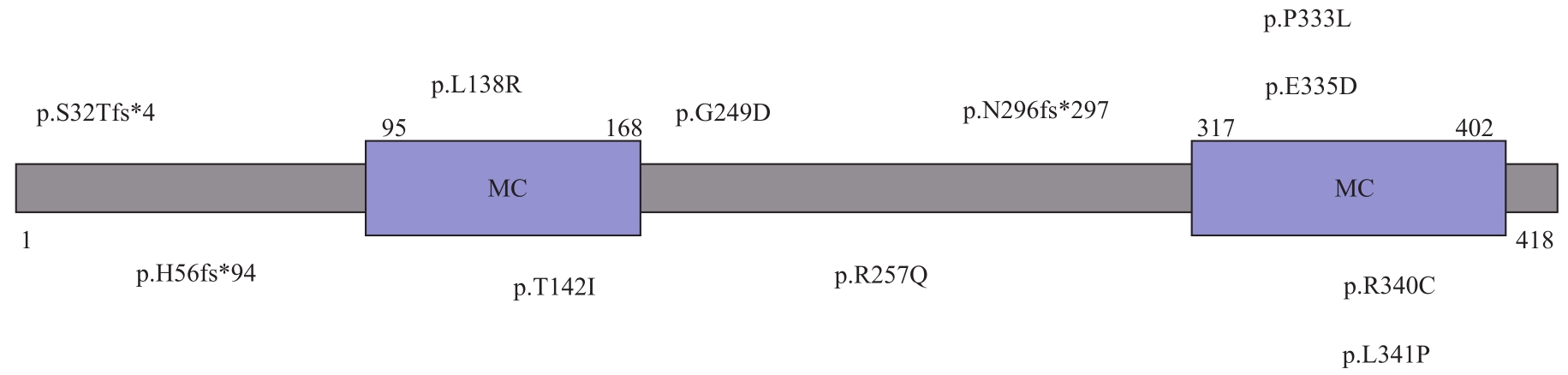
图3 SLC25A46的结构示意图及突变分布Note: MC—mitochondrial carrier domain.
Fig 3 Structure diagram and mutation distribution of SLC25A46
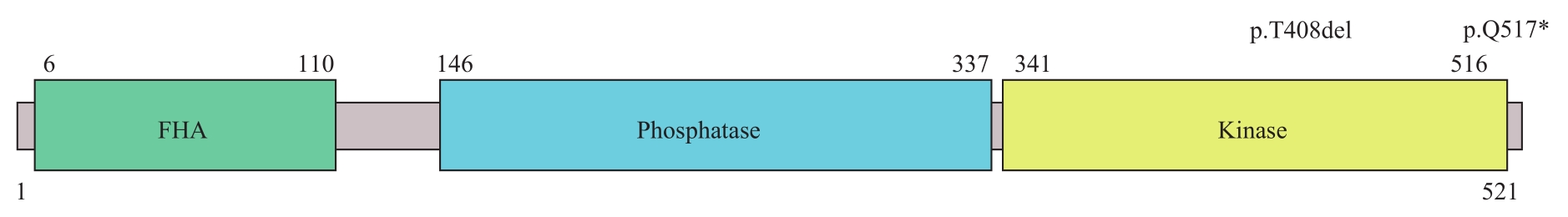
图4 PNKP的结构示意图及突变分布Note: FHA—forkhead associated domain.
Fig 4 Structure diagram and mutation distribution of PNKP
| 1 | LAURÁ M, PIPIS M, ROSSOR A M, et al. Charcot-Marie-Tooth disease and related disorders: an evolving landscape[J]. Curr Opin Neurol, 2019, 32(5): 641-650. |
| 2 | ROSSOR A M, CARR A S, DEVINE H, et al. Peripheral neuropathy in complex inherited diseases: an approach to diagnosis[J]. J Neurol Neurosurg Psychiatry, 2017, 88(10): 846-863. |
| 3 | MERSIYANOVA I V, PEREPELOV A V, POLYAKOV A V, et al. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene[J]. Am J Hum Genet, 2000, 67(1): 37-46. |
| 4 | STONE E J, KOLB S J, BROWN A. A review and analysis of the clinical literature on Charcot-Marie-Tooth disease caused by mutations in neurofilament protein L[J]. Cytoskeleton (Hoboken), 2021, 78(3): 97-110. |
| 5 | ABE A, NUMAKURA C, SAITO K, et al. Neurofilament light chain polypeptide gene mutations in Charcot-Marie-Tooth disease: nonsense mutation probably causes a recessive phenotype[J]. J Hum Genet, 2009, 54(2): 94-97. |
| 6 | ANTUNES L, FRASQUILHO S, OSTASZEWSKI M, et al. Similar α-synuclein staining in the colon mucosa in patients with Parkinson′s disease and controls[J]. Mov Disord, 2016, 31(10): 1567-1570. |
| 7 | ZHAI J B, LIN H, JULIEN J P, et al. Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1[J]. Hum Mol Genet, 2007, 16(24): 3103-3116. |
| 8 | MARTIN M, IYADURAI S J, GASSMAN A, et al. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport[J]. Mol Biol Cell, 1999, 10(11): 3717-3728. |
| 9 | YUM S W, ZHANG J X, MO K T, et al. A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy[J]. Ann Neurol, 2009, 66(6): 759-770. |
| 10 | LERAT J, MAGDELAINE C, BEAUVAIS-DZUGAN H, et al. A novel pathogenic variant of NEFL responsible for deafness associated with peripheral neuropathy discovered through next-generation sequencing and review of the literature[J]. J Peripher Nerv Syst, 2019, 24(1): 139-144. |
| 11 | GEORGIOU D M, ZIDAR J, KOROSEC M, et al. A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family[J]. Neurogenetics, 2002, 4(2): 93-96. |
| 12 | FABRIZI G M, CAVALLARO T, ANGIARI C, et al. Giant axon and neurofilament accumulation in Charcot-Marie-Tooth disease type 2E[J]. Neurology, 2004, 62(8): 1429-1431. |
| 13 | KIM H J, KIM S B, KIM H S, et al. Phenotypic heterogeneity in patients with NEFL-related Charcot-Marie-Tooth disease[J]. Mol Genet Genomic Med, 2022, 10(2): e1870. |
| 14 | LIKAR T, HASANHODŽIĆ M, TERAN N, et al. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss[J]. PLoS One, 2018, 13(1): e0188578. |
| 15 | PISCIOTTA C, BAI Y H, BRENNAN K M, et al. Reduced neurofilament expression in cutaneous nerve fibers of patients with CMT2E[J]. Neurology, 2015, 85(3): 228-234. |
| 16 | AGRAWAL P B, JOSHI M, MARINAKIS N S, et al. Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings[J]. JAMA Neurol, 2014, 71(11): 1413-1420. |
| 17 | HORGA A, LAURÀ M, JAUNMUKTANE Z, et al. Genetic and clinical characteristics of NEFL-related Charcot-Marie-Tooth disease[J]. J Neurol Neurosurg Psychiatry, 2017, 88(7): 575-585. |
| 18 | DOPPLER K, KUNSTMANN E, KRÜGER S, et al. Painful Charcot-Marie-Tooth neuropathy type 2E/1F due to a novel NEFL mutation[J]. Muscle Nerve, 2017, 55(5): 752-755. |
| 19 | SMITH S M, JENKINSON M, JOHANSEN-BERG H, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data[J]. NeuroImage, 2006, 31(4): 1487-1505. |
| 20 | HWANG S, PARK C H, KIM R E, et al. Cerebellar white matter abnormalities in Charcot-Marie-Tooth Disease: a combined volumetry and diffusion tensor imaging analysis[J]. J Clin Med, 2021, 10(21): 4945. |
| 21 | ANDO M, OKAMOTO Y, YOSHIMURA A, et al. Clinical and mutational spectrum of Charcot-Marie-Tooth disease type 2Z caused by MORC2 variants in Japan[J]. Eur J Neurol, 2017, 24(10): 1274-1282. |
| 22 | PAREYSON D, SAVERI P, PISCIOTTA C. New developments in Charcot-Marie-Tooth neuropathy and related diseases[J]. Curr Opin Neurol, 2017, 30(5): 471-480. |
| 23 | GUILLEN SACOTO M J, TCHASOVNIKAROVA I A, TORTI E, et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism[J]. Am J Hum Genet, 2020, 107(2): 352-363. |
| 24 | SANCHO P, BARTESAGHI L, MIOSSEC O, et al. Characterization of molecular mechanisms underlying the axonal Charcot-Marie-Tooth neuropathy caused by MORC2 mutations[J]. Hum Mol Genet, 2019, 28(10): 1629-1644. |
| 25 | SEVILLA T, LUPO V, MARTÍNEZ-RUBIO D, et al. Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease[J]. Brain, 2016, 139(Pt 1): 62-72. |
| 26 | WANG G L, WANG C Y, CAI X Z, et al. Identification and expression analysis of a novel CW-type zinc finger protein MORC2 in cancer cells[J]. Anat Rec (Hoboken), 2010, 293(6): 1002-1009. |
| 27 | TCHASOVNIKAROVA I A, TIMMS R T, DOUSE C H, et al. Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2[J]. Nat Genet, 2017, 49(7): 1035-1044. |
| 28 | LI D Q, NAIR S S, OHSHIRO K, et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response[J]. Cell Rep, 2012, 2(6): 1657-1669. |
| 29 | DOUSE C H, BLOOR S, LIU Y C, et al. Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms[J]. Nat Commun, 2018, 9(1): 651. |
| 30 | SÁNCHEZ-SOLANA B, LI D Q, KUMAR R. Cytosolic functions of MORC2 in lipogenesis and adipogenesis[J]. Biochim Biophys Acta, 2014, 1843(2): 316-326. |
| 31 | SHAO Y G, LI Y, ZHANG J, et al. Involvement of histone deacetylation in MORC2-mediated down-regulation of carbonic anhydrase Ⅸ[J]. Nucleic Acids Res, 2010, 38(9): 2813-2824. |
| 32 | CESTRA G, TOOMRE D, CHANG S, et al. The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton[J]. Proc Natl Acad Sci U S A, 2005, 102(5): 1731-1736. |
| 33 | ZANNI G, NARDELLA M, BARRESI S, et al. De novo p.T362R mutation in MORC2 causes early onset cerebellar ataxia, axonal polyneuropathy and nocturnal hypoventilation[J]. Brain, 2017, 140(6): e34. |
| 34 | SCHOTTMANN G, WAGNER C, SEIFERT F, et al. MORC2 mutation causes severe spinal muscular atrophy-phenotype, cerebellar atrophy, and diaphragmatic paralysis[J]. Brain, 2016, 139(Pt 12): e70. |
| 35 | HYUN Y S, HONG Y B, CHOI B O, et al. Clinico-genetics in Korean Charcot-Marie-Tooth disease type 2Z with MORC2 mutations[J]. Brain, 2016, 139(Pt 7): e40. |
| 36 | SIVERA R, LUPO V, FRASQUET M, et al. Charcot-Marie-Tooth disease due to MORC2 mutations in Spain[J]. Eur J Neurol, 2021, 28(9): 3001-3011. |
| 37 | RAJU S, MEDARAMETLA S, BORAIAH N. Dystonia and hereditary motor sensory neuropathy 6B due to SLC25A46 gene mutations[J]. Mov Disord Clin Pract, 2021, 8(3): 480-482. |
| 38 | ABRAMS A J, HUFNAGEL R B, REBELO A, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder[J]. Nat Genet, 2015, 47(8): 926-932. |
| 39 | PERIVOLIDI V I, VIOLITZI F, IOANNIDOU E, et al. Proteomic identification of the SLC25A46 interactome in transgenic mice expressing SLC25A46-FLAG[J]. J Proteome Res, 2022, 21(2): 375-394. |
| 40 | WAN J J, STEFFEN J, YOURSHAW M, et al. Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia[J]. Brain, 2016, 139(11): 2877-2890. |
| 41 | GAO L, WANG M, LIAO L F, et al. A Slc25a46 mouse model simulating age-associated motor deficit, redox imbalance, and mitochondria dysfunction[J]. J Gerontol A Biol Sci Med Sci, 2021, 76(3): 440-447. |
| 42 | LI Z, PENG Y Y, HUFNAGEL R B, et al. Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice[J]. Hum Mol Genet, 2017, 26(19): 3776-3791. |
| 43 | NGUYEN M, BOESTEN I, HELLEBREKERS D M, et al. Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder[J]. Clin Genet, 2017, 91(1): 121-125. |
| 44 | ABRAMS A J, FONTANESI F, TAN N B L, et al. Insights into the genotype-phenotype correlation and molecular function of SLC25A46[J]. Hum Mutat, 2018, 39(12): 1995-2007. |
| 45 | BRAUNISCH M C, GALLWITZ H, ABICHT A, et al. Extension of the phenotype of biallelic loss-of-function mutations in SLC25A46 to the severe form of pontocerebellar hypoplasia type Ⅰ[J]. Clin Genet, 2018, 93(2): 255-265. |
| 46 | CHARLESWORTH G, BALINT B, MENCACCI N E, et al. SLC25A46 mutations underlie progressive myoclonic ataxia with optic atrophy and neuropathy[J]. Mov Disord, 2016, 31(8): 1249-1251. |
| 47 | LEAL A, BOGANTES-LEDEZMA S, EKICI A B, et al. The polynucleotide kinase 3′-phosphatase gene (PNKP) is involved in Charcot-Marie-Tooth disease (CMT2B2) previously related to MED25[J]. Neurogenetics, 2018, 19(4): 215-225. |
| 48 | LEAL A, MORERA B, DEL VALLEG, et al. A second locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 19q13.3[J]. Am J Hum Genet, 2001, 68(1): 269-274. |
| 49 | JILANI A, RAMOTAR D, SLACK C, et al. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage[J]. J Biol Chem, 1999, 274(34): 24176-24186. |
| 50 | BERNSTEIN N K, WILLIAMS R S, RAKOVSZKY M L, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase[J]. Mol Cell, 2005, 17(5): 657-670. |
| 51 | TSUKADA K, MATSUMOTO Y, SHIMADA M. Linker region is required for efficient nuclear localization of polynucleotide kinase phosphatase[J]. PLoS One, 2020, 15(9): e0239404. |
| 52 | MCKINNON P J. DNA repair deficiency and neurological disease[J]. Nat Rev Neurosci, 2009, 10(2): 100-112. |
| 53 | PEDROSO J L, ROCHA C R, MACEDO-SOUZA L I, et al. Mutation in PNKP presenting initially as axonal Charcot-Marie-Tooth disease[J]. Neurol Genet, 2015, 1(4): e30. |
| 54 | BERGHOFF C, BERGHOFF M, LEAL A, et al. Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2B) that maps to chromosome 19q13.3[J]. Neuromuscul Disord, 2004, 14(5): 301-306. |
| 55 | BRAS J, ALONSO I, BARBOT C, et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4[J]. Am J Hum Genet, 2015, 96(3): 474-479. |
| 56 | DJORDJEVIC D, PINARD M, GAUTHIER M S, et al. De novo variants in POLR3B cause ataxia, spasticity, and demyelinating neuropathy[J]. Am J Hum Genet, 2022, 109(4): 759-763. |
| 57 | HYUN Y S, PARK H J, HEO S H, et al. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy[J]. Clin Genet, 2014, 86(6): 592-594. |
| 58 | CORTESE A, SIMONE R, SULLIVAN R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia[J]. Nat Genet, 2019, 51(4): 649-658. |
| 59 | 李资益, 田沃土, 沈隽逸, 等. 非典型Charlevoix-Saguenay常染色体隐性遗传痉挛性共济失调二家系[J]. 中国神经精神疾病杂志, 2021, 47(8): 509-512. |
| LI Z Y, TIAN W T, SHEN J Y, et al. Atypical autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of two families[J]. Chinese Journal of Nervous and Mental Diseases, 2021, 47(8): 509-512. | |
| 60 | ECHANIZ-LAGUNA A, GHEZZI D, CHASSAGNE M, et al. SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease[J]. Neurology, 2013, 81(17): 1523-1530. |
| [1] | 朱啸巍, 詹飞霞, 张超, 刘时华, 钟平, 曹立, 栾兴华. PMP22基因相关性周围神经病的遗传学和临床特点分析[J]. 上海交通大学学报(医学版), 2022, 42(5): 609-616. |
| [2] | 谢雨婕, 谢利娟, 朱天闻, 王依闻. 白介素-10受体A基因突变致新生儿极早发炎性肠病2例[J]. 上海交通大学学报(医学版), 2021, 41(3): 409-412. |
| [3] | 汪群峰, 曹立, 栾兴华. 周围神经病评分量表的临床应用综述[J]. 上海交通大学学报(医学版), 2021, 41(11): 1518-1523. |
| [4] | 朱之星1, 2,纪 伟3,顾坚磊1, 4,吕 晖1, 2, 4,田国力3. 葡萄糖 -6- 磷酸脱氢酶基因4种突变及蛋白结构分析[J]. 上海交通大学学报(医学版), 2020, 40(12): 1571-1578. |
| [5] | 刘思捷 1,李婷婷 1,陈笋 1,李奋 2,孙锟 1,徐让 3. CITED2基因在内脏反位患者中的突变分析[J]. 上海交通大学学报(医学版), 2019, 39(5): 500-. |
| [6] | 严天奇 1,陈立伟 2,朱勇梅 2,李剑峰 2,代雨婷 2, 3,崔舒雅 2,姜璐 2,陈冰 2,黄金艳 2. 急性淋巴细胞白血病基因融合与突变知识库的构建[J]. 上海交通大学学报(医学版), 2018, 38(9): 1027-. |
| [7] | 李青丽,文君,闵雪洁,赵丽,赵小平. 异柠檬酸脱氢酶基因突变在急性髓细胞白血病发生中的作用[J]. 上海交通大学学报(医学版), 2018, 38(8): 960-. |
| [8] | 王炳华 1,许无恨 2,汤晓君 2,兰小平 2. 中国汉族人群KRT9基因突变分析[J]. 上海交通大学学报(医学版), 2018, 38(12): 1425-. |
| [9] | 陈珺珏,田琳璐,韦严,亢晓丽. X染色体连锁显性遗传婴儿眼球震颤家系的临床特征研究[J]. 上海交通大学学报(医学版), 2018, 38(11): 1355-. |
| [10] | 洪莎 1,赵冬莹 1,谢利娟 1,常国营 2,刘晓青 3,朱天闻 1. 2个中国汉族假肥大型进行性肌营养不良家系分析[J]. 上海交通大学学报(医学版), 2018, 38(10): 1223-. |
| [11] | 侯悦,李少波,师晓琴,傅国辉,伍均. 结直肠癌KRAS 、NRAS 和BRAF 基因突变与临床病理特征的回顾性分析[J]. 上海交通大学学报(医学版), 2018, 38(1): 4-. |
| [12] | 石晓东,戴钰俊,王月英. DNMT3A 在血液肿瘤中的作用#br#[J]. 上海交通大学学报(医学版), 2017, 37(9): 1276-. |
| [13] | 顾本宏,朱晓斌,朱子珏,田汝辉,李朋,智二磊,姚晨成,王洪,陈慧兴,万众,黄煜华,何祖平,李铮 . EDA基因新剪切突变导致X连锁少汗性外胚层发育不良[J]. 上海交通大学学报(医学版), 2017, 37(3): 288-. |
| [14] | 李剑峰,严天奇,崔博文,孔杰,王舒,陈冰,黄金艳 . 基于基因 Panel 测序数据的分析方法[J]. 上海交通大学学报(医学版), 2017, 37(11): 1575-. |
| [15] | 王 舒,陈 冰. 急性髓细胞白血病的分子分型[J]. 上海交通大学学报(医学版), 2016, 36(8): 1223-. |
| 阅读次数 | ||||||||||||||||||||||||||||||||||||||||||||||||||
|
全文 657
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
摘要 436
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||