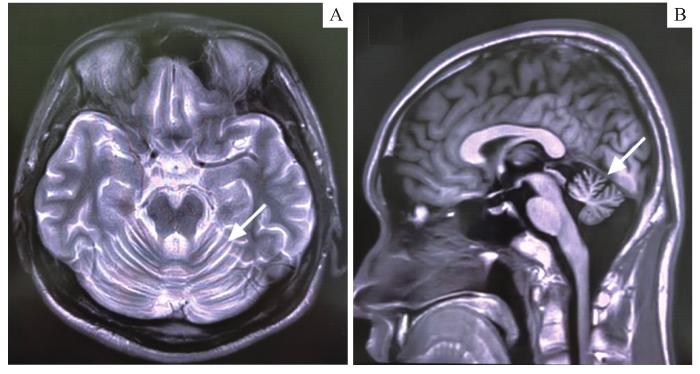
Spinocerebellar ataxia (SCA) is a rare autosomal neurodegenerative disease. SCA28 is a rare subtype, caused by heterozygous mutation of the pathogenic gene AFG3L2. The clinical features usually include slowly progressive gait and limb ataxia, dysarthria, hyperreflexia of the lower limbs, gaze-evoked nystagmus, ptosis, ophthalmoplegia, decreased ankle reflex, Parkinsonism, dystonia and cognitive impairment. In this paper, a case of SCA28 with orthostatic tremor is reported. The gene detection showed that there was a missense mutation of c. 2098G>A in the AFG3L2 gene of the patient. The clinical symptoms and pathogenic gene mutations of 79 cases of SCA28 type in the previous relevant literature are summarized to strengthen the understanding and clinical diagnosis of the disease.
WANG Qunfeng, LIU Shihua. A case of spinocerebellar ataxia type 28 with orthostatic tremor. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2023, 43(4): 514-518 doi:10.3969/j.issn.1674-8115.2023.04.016
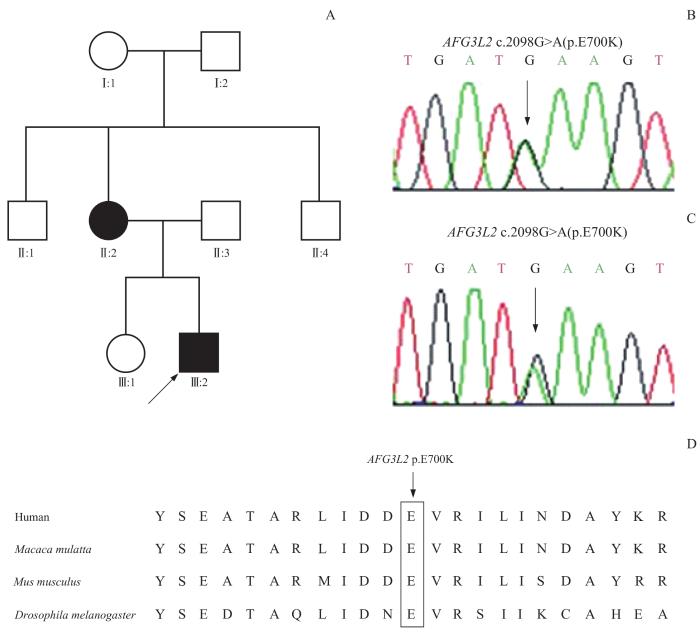
Note:A. Family diagram of the proband. B. Missense mutation c.2098G>A (p.E700K) in the proband. C. Missense mutation c.2098G>A (p.E700K) in the proband's mother. D. Conservative analysis of different species.
Fig 2
Pedigree of the proband and AFG3L2 Sanger sequencing diagram
1.3 基因测序
抽取先证者外周血样本,采用重复引物PCR(repeat-primed PCR,RP-PCR)和毛细管电泳技术筛查和排除了SCA1、2、3、6、7、8、10、12、17、36,以及齿状核红核苍白球路易体萎缩症(dentatorubrar-pallidoluysian atrophy,DRPLA)和弗里德赖希共济失调(Friedreich ataxia,FRDA)的动态突变。为了明确先证者的致病基因,对先证者外周血DNA进行全外显子组基因测序,结果显示AFG3L2基因(chr18:12337417)存在c.2098G>A(p.E700K)错义突变(图2B)。筛选先证者和其母亲的AFG3L2基因进行Sanger测序和家系共分离,结果显示AFG3L2基因是致病基因(图2C)。突变位点在不同物种间是非常保守的(图2D)。根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics,ACMG)指南,变异c.2098G>A(p.E700K)被评为“可能致病”。结合患者临床特征和基因检测结果,诊断该病为SCA28。先证者接受了对症支持药物治疗,其母亲未接受任何治疗。2例患者共济失调症状持续恶化。
The paper was designed by LIU Shihua. The clinical data of the patient was collected by WANG Qunfeng and LIU Shihua. The manuscript was drafted and revised by WANG Qunfeng. Both authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
Both authors disclose no relevant conflict of interests.
PAULSON H L, SHAKKOTTAI V G, CLARK H B, et al. Polyglutamine spinocerebellar ataxias:from genes to potential treatments[J]. Nat Rev Neurosci, 2017, 18(10): 613-626.
TULLI S, DEL BONDIO A, BADERNA V, et al. Pathogenic variants in the AFG3L2 proteolytic domain cause SCA28 through haploinsufficiency and proteostatic stress-driven OMA1 activation[J]. J Med Genet, 2019, 56(8): 499-511.
RUANO L, MELO C, SILVA M C, et al. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies[J]. Neuroepidemiology, 2014, 42(3): 174-183.
CAGNOLI C, MARIOTTI C, TARONI F, et al. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-Q11.2[J]. Brain, 2006, 129(1): 235-242.
DI BELLA D, LAZZARO F, BRUSCO A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28[J]. Nat Genet, 2010, 42(4): 313-321.
EDENER U, WÖLLNER J, HEHR U, et al. Early onset and slow progression of SCA28, a rare dominant ataxia in a large four-generation family with a novel AFG3L2 mutation[J]. Eur J Hum Genet, 2010, 18(8): 965-968.
MARIOTTI C, BRUSCO A, DI BELLA D, et al. Spinocerebellar ataxia type 28: a novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis[J]. Cerebellum, 2008, 7(2): 184-188.
CAGNOLI C, STEVANIN G, BRUSSINO A, et al. Missense mutations in the AFG3L2 proteolytic domain account for 1.5% of European autosomal dominant cerebellar ataxias[J]. Hum Mutat, 2010, 31(10): 1117-1124.
LIU X, WANG L, CHEN J, et al. Spinocerebellar ataxia type 28 in a Chinese pedigree: a case report and literature review[J]. Medicine (Baltimore), 2021, 100(50): e28008.
CHIANG H L, FUH J L, TSAI Y S, et al. Expanding the phenotype of AFG3L2 mutations: late-onset autosomal recessive spinocerebellar ataxia[J]. J Neurol Sci, 2021, 428: 117600.
CALANDRA C R, BUDA G, VISHNOPOLSKA S A, et al. Spastic ataxia with eye-of-the-tiger-like sign in 4 siblings due to novel compound heterozygous AFG3L2 mutation[J]. Parkinsonism Relat Disord, 2020, 73: 52-54.
TUNC S, DULOVIC-MAHLOW M, BAUMANN H, et al. Spinocerebellar ataxia type 28-phenotypic and molecular characterization of a family with heterozygous and compound-heterozygous mutations in AFG3L2[J]. Cerebellum, 2019, 18(4): 817-822.
SZPISJAK L, NEMETH V L, SZEPFALUSI N, et al. Neurocognitive characterization of an SCA28 family caused by a novel AFG3L2 gene mutation[J]. Cerebellum, 2017, 16(5/6): 979-985.
POLITI L S, BIANCHI MARZOLI S, GODI C, et al. MRI evidence of cerebellar and extraocular muscle atrophy differently contributing to eye movement abnormalities in SCA2 and SCA28 diseases[J]. Invest Ophthalmol Vis Sci, 2016, 57(6): 2714-2720.
ZÜHLKE C, MIKAT B, TIMMANN D, et al. Spinocerebellar ataxia 28: a novel AFG3L2 mutation in a German family with young onset, slow progression and saccadic slowing[J]. Cerebellum Ataxias, 2015, 2: 19.
QU J, WU C K, ZUZUÁRREGUI J R, et al. A novel AFG3L2 mutation in a Somalian patient with spinocerebellar ataxia type 28[J]. J Neurol Sci, 2015, 358(1/2): 530-531.
SMETS K, DECONINCK T, BAETS J, et al. Partial deletion of AFG3L2 causing spinocerebellar ataxia type 28[J]. Neurology, 2014, 82(23): 2092-2100.
MUSOVA Z, KAISEROVA M, KRIEGOVA E, et al. A novel frameshift mutation in the AFG3L2 gene in a patient with spinocerebellar ataxia[J]. Cerebellum, 2014, 13(3): 331-337.
LÖBBE A M, KANG J S, HILKER R, et al. A novel missense mutation in AFG3L2 associated with late onset and slow progression of spinocerebellar ataxia type 28[J]. J Mol Neurosci, 2014, 52(4): 493-496.



{kind=link}
{kind=link}
{kind=link}
{kind=link}