Progress in the regulatory mechanisms of mandibular condylar development and deformity
LIU Jingyi,, XU Hongyuan, DAI Qinggang, JIANG Lingyong,
Department of Oral and Craniomaxillofacial Surgery, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine; College of Stomatology, Shanghai Jiao Tong University; National Center for Stomatology; National Clinical Research Center for Oral Diseases; Shanghai Key Laboratory of Stomatology; Shanghai Research Institute of Stomatology, Shanghai 200011, China
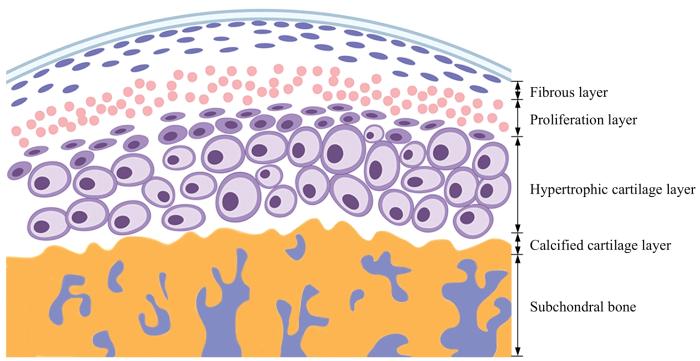
The temporomandibular joint is the only joint structure within the craniofacial skeletal system, responsible for performing functions related to opening and closing mouth movements, such as chewing, speaking, and facial expression in daily life. The condyle of the mandible, as a vital component of the temporomandibular joint, originates from the mandibular process formed by the first gill arch and is the key growth center at the end of the mandibular ramus. Condyle is composed of a layer of cartilage as its surface and subchondral bone below, exhibiting unique biological processes during its growth and development. In the articular fossa, the functional movement of the condyle depends on its normal physiological and anatomical structure, which plays a crucial role in establishing occlusion and shaping facial features. Abnormal growth and development can lead to the occurrence of condylar deformities, which affect the vertical height of the patient's maxillofacial region and ultimately lead to secondary skeletal class Ⅱ or Ⅲ craniofacial deformities. During the process of growth and development, the condyle is subject to complex signal regulation. In recent years, with in-depth research on the temporomandibular joint, researchers have begun to discuss the regulatory mechanisms of condyle growth and development from the perspectives of gene expression and molecular level, in order to explain the causes of temporomandibular joint diseases and condylar deformities. This article provides a review on the growth process and structure of condyle, classification and pathological manifestations of condylar deformities, and related regulatory mechanisms of the growth and development of condyle, as well as pathogenesis of condylar deformities. The aim of this article is to provide research ideas for temporomandibular joint diseases and craniofacial malformations caused by abnormal development of the mandibular condyle in clinical practice.
Keywords:condyle
;
growth and development
;
deformity
;
regulatory mechanism
;
bone remodeling
LIU Jingyi, XU Hongyuan, DAI Qinggang, JIANG Lingyong. Progress in the regulatory mechanisms of mandibular condylar development and deformity. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2024, 44(8): 951-958 doi:10.3969/j.issn.1674-8115.2024.08.003
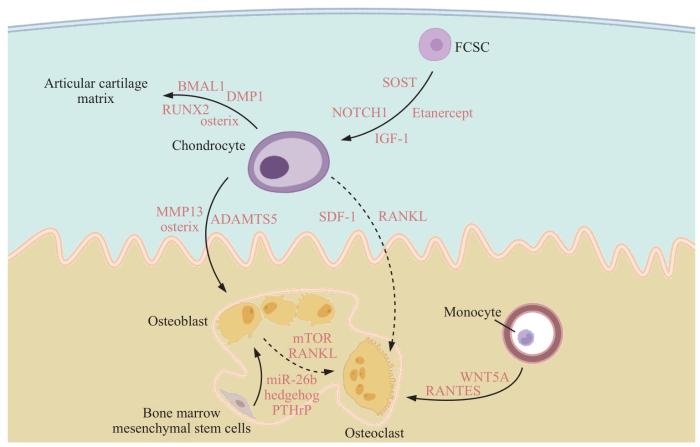
髁突的成骨细胞有2个来源,分别是骨髓间充质干细胞分化和肥大软骨细胞的转分化,两者都对软骨下骨的骨形成起到重要作用。ZHANG等[36]发现,甲状旁腺素相关肽(parathyroid hormone-related peptide,PTHrP)能够改善软骨下骨的骨髓微环境,髁突软骨下骨髓间充质干细胞的甲状旁腺素相关肽/甲状旁腺激素受体1(parathyroid hormone-related peptide/parathyroid hormone receptor 1,PTHrP/PTH1R)通路的激活会抑制分化过程中的转化生长因子β(transforming growth factor-β,TGF-β)信号转导,促进其向成骨细胞分化。LEI等[37]通过标志物神经胶质瘤致病基因同源物1(glioma-associated oncogene homolog 1,GLI1)鉴定出髁突软骨下骨间充质干细胞来源的成骨祖细胞,并发现Hedgehog/GLI1信号通路的激活会导致异常的骨重塑,靶向Gli1+成骨祖细胞中的Hedgehog信号可调节软骨下骨区的骨稳态。miRNA-26b也被证明对软骨下骨骨髓间充质干细胞的成骨分化具有调控作用[38],但是缺乏相关结果验证其在小鼠髁突生长发育中的作用。除骨髓间充质干细胞外,软骨下骨的形成也和软骨细胞密切相关。JING等[39]研究显示,成骨细胞分化和骨矿化相关因子osterix与软骨区软骨形成和软骨下骨区的骨形成密切相关,在软骨细胞中敲除osterix会导致软骨层中肥大细胞累积和软骨基质钙化受阻,说明髁突软骨下骨的骨形成和软骨细胞的分化有关,而不是彼此独立的过程。随后JING等[9]通过谱系示踪首先报道了髁突软骨中的肥大软骨细胞向软骨下骨成骨细胞转分化的现象。ROGERS-DECOTES等[40]发现,细胞外金属基质蛋白酶ADAMTS5(ADAM metallopeptidase with thrombospondin type 1 motif 5)有利于肥大细胞向成骨细胞的转分化,该酶的敲除抑制了肥大细胞在向成骨细胞转分化过程中所依赖的周围软骨基质的降解,导致软骨下骨成骨细胞数量减少,同时骨量减少、骨小梁厚度降低。以上结果显示,骨髓间充质干细胞分化和肥大细胞转分化受阻均会破坏成骨细胞的形成,影响生长发育过程中的正常骨形成。
髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞。在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41]。在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能。YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少。此外,外界的机械应力也能激活软骨下骨中破骨细胞分化。KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子。TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成。FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失。HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏。TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成。在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱。综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育。
髁突肥大被认为是软骨细胞增殖异常增加及凋亡减弱的结果,而髁突发育不良则与之相反。CHEN等[48]分离髁突肥大患者的髁突软骨细胞,发现与正常软骨细胞相比,IGF-1的表达水平明显升高,高水平IGF-1通过MAPK/ERK(mitogen activated protein kinase/extracellular signal-regulated kinase)信号通路促进软骨细胞的增殖,同时增加软骨基质的合成,诱导髁突肥大的发生。在该研究结果的基础上,CAO等[49]研究发现miR-15b在髁突肥大患者的髁突软骨细胞中的表达显著降低。IGF-1、IGF-1受体及抗凋亡因子BCL2作为miR-15b的直接作用靶点,因miR-15b的抑制作用降低而出现表达升高,导致软骨细胞增殖异常增加,凋亡减弱,促进髁突肥大的发生。
髁突发育不良的发生与髁突肥大相反,为软骨细胞凋亡异常增加,而增殖分化受阻的结果。WEN等[50]发现转铁蛋白(transferrin,TF)能够保护软骨细胞免受缺氧诱导的细胞凋亡,并且通过ULK1/ATG16L1(Unc-51 like autophagy activating kinase 1/autophagy related 16 like 1)信号轴诱导自噬,促进软骨细胞的分化。髁突发育不良患者血清中检测到TF水平降低,导致软骨细胞凋亡增加、增殖与分化受阻,最终引发疾病。此外,BIOSSE DUPLAN等[51]研究显示,在侏儒症患者中常见的成纤维细胞生长因子受体3(fibroblast growth factor receptor 3,FGFR3)功能获得性突变引发的受体过度激活会导致胚胎在下颌形成过程中麦氏软骨和髁突软骨的软骨细胞增殖分化能力缺陷,从而引发下颌骨发育不全及髁突畸形的发生。
LIU Jingyi was responsible for literature collection and review writing. XU Hongyuan, DAI Qinggang, JIANG Lingyong were responsible for guiding and reviewing the paper. All the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
All authors disclose no relevant conflict of interests.
KAYIPMAZ S, AKÇAY S, SEZGIN Ö S, et al. Trabecular structural changes in the mandibular condyle caused by degenerative osteoarthritis: a comparative study by cone-beam computed tomography imaging[J]. Oral Radiol, 2019, 35(1): 51-58.
TANG G H, RABIE A B. Runx2 regulates endochondral ossification in condyle during mandibular advancement[J]. J Dent Res, 2005, 84(2): 166-171.
HIROUCHI H, KITAMURA K, YAMAMOTO M, et al. Developmental characteristics of secondary cartilage in the mandibular condyle and sphenoid bone in mice[J]. Arch Oral Biol, 2018, 89: 84-92.
XU X J, ZHANG Y J, ZHANG J, et al. Zonal interdependence in the temporomandibular joint cartilage[J]. FASEB J, 2023, 37(4): e22888.
ZHU Q Q, TAN M Y, WANG C N, et al. Single-cell RNA sequencing analysis of the temporomandibular joint condyle in 3 and 4-month-old human embryos[J]. Cell Biosci, 2023, 13(1): 130.
LIU C, JIA Y, YANG S S, et al. Characteristics of the growth, development and microarchitecture of condyle subchondral bone in rats[J]. Chinese Journal of Tissue Engineering Research, 2022, 26(32): 5162-5166.
KANEYAMA K, SEGAMI N, HATTA T. Congenital deformities and developmental abnormalities of the mandibular condyle in the temporomandibular joint[J]. Congenit Anom, 2008, 48(3): 118-125.
GALEA C J, DASHOW J E, WOERNER J E. Congenital abnormalities of the temporomandibular joint[J]. Oral Maxillofac Surg Clin North Am, 2018, 30(1): 71-82.
CHOUINARD A F, KABAN L B, PEACOCK Z S. Acquired abnormalities of the temporomandibular joint[J]. Oral Maxillofac Surg Clin North Am, 2018, 30(1): 83-96.
YU J S, YANG T, DAI J W, et al. Histopathological features of condylar hyperplasia and condylar Osteochondroma: a comparison study[J]. Orphanet J Rare Dis, 2019, 14(1): 293.
VÁSQUEZ B, OLATE S, CANTÍN M, et al. Histomorphometric analysis of unilateral condylar hyperplasia in the temporomandibular joint: the value of the condylar layer and cartilage island[J]. Int J Oral Maxillofac Surg, 2017, 46(7): 861-866.
CHEN Y X, HUANG Q, ZHANG W Y, et al. Animal experiment on cooperation between Shh and IGF-1 in promoting mandibular cartilage overgrowth[J]. Journal of Oral Science Research, 2018, 34 (8): 866-869.
LI X H, LIANG W N, YE H Z, et al. Overexpression of Shox2 leads to congenital dysplasia of the temporomandibular joint in mice[J]. Int J Mol Sci, 2014, 15(8): 13135-13150.
SUN L, ZHAO J, WANG H, et al. Mechanical stress promotes matrix synthesis of mandibular condylar cartilage via the RKIP-ERK pathway[J]. J Mol Histol, 2017, 48(5/6): 437-446.
MARTÍN A E, DEL R PANI M, HOLGADO N R, et al. Facial development disorders due to inhibition to endochondral ossification of mandibular condyle process caused by malnutrition[J]. Angle Orthod, 2014, 84(3): 473-478.
BI R Y, LI Q L, LI H H, et al. Divergent chondro/osteogenic transduction laws of fibrocartilage stem cell drive temporomandibular joint osteoarthritis in growing mice[J]. Int J Oral Sci, 2023, 15(1): 36.
RUSCITTO A, CHEN P, TOSA I, et al. Lgr5-expressing secretory cells form a Wnt inhibitory niche in cartilage critical for chondrocyte identity[J]. Cell Stem Cell, 2023, 30(9): 1179-1198.e7.
RUSCITTO A, SCARPA V, MOREL M, et al. Notch regulates fibrocartilage stem cell fate and is upregulated in inflammatory TMJ arthritis[J]. J Dent Res, 2020, 99(10): 1174-1181.
BI R Y, LUO X T, LI Q L, et al. Igf1 regulates fibrocartilage stem cells, cartilage growth, and homeostasis in the temporomandibular joint of mice[J]. J Bone Miner Res, 2023, 38(4): 556-567.
JOSHI A S, HATCH N E, HAYAMI T, et al. IGF-1 TMJ injections enhance mandibular growth and bone quality in juvenile rats[J]. Orthod Craniofac Res, 2022, 25(2): 183-191.
KURIO N, SAUNDERS C, BECHTOLD T E, et al. Roles of Ihh signaling in chondroprogenitor function in postnatal condylar cartilage[J]. Matrix Biol, 2018, 67: 15-31.
LIAO L F, ZHANG S X, ZHOU G Q, et al. Deletion of Runx2 in condylar chondrocytes disrupts TMJ tissue homeostasis[J]. J Cell Physiol, 2019, 234(4): 3436-3444.
GE C, MOHAMED F, BINRAYES A, et al. Selective role of discoidin domain receptor 2 in murine temporomandibular joint development and aging[J]. J Dent Res, 2018, 97(3): 321-328.
YANG J L, XU Y F, XUE X, et al. MicroRNA-26b regulates BMSC osteogenic differentiation of TMJ subchondral bone through β-catenin in osteoarthritis[J]. Bone, 2022, 162: 116448.
JING J, HINTON R J, JING Y, et al. Osterix couples chondrogenesis and osteogenesis in post-natal condylar growth[J]. J Dent Res, 2014, 93(10): 1014-1021.
ROGERS-DECOTES A W, PORTO S C, DUPUIS L E, et al. ADAMTS5 is required for normal trabeculated bone development in the mandibular condyle[J]. Osteoarthritis Cartilage, 2021, 29(4): 547-557.
OYHANART S R, ESCUDERO N D, MANDALUNIS P M. Effect of alendronate on the mandible and long bones: an experimental study in vivo[J]. Pediatr Res, 2015, 78(6): 618-625.
TIAN Y H, CHEN J B, YAN X, et al. Overloaded orthopedic force induces condylar subchondral bone absorption by stimulating rat mesenchymal stem cells differentiating into osteoclasts via mTOR-regulated RANKL/OPG secretion in osteoblasts[J]. Stem Cells Dev, 2021, 30(1): 29-38.
FENG S Y, LEI J, LI Y X, et al. Increased joint loading induces subchondral bone loss of the temporomandibular joint via the RANTES-CCRs-Akt2 axis[J]. JCI Insight, 2022, 7(21): e158874.
HE L H, XIAO E, DUAN D H, et al. Osteoclast deficiency contributes to temporomandibular joint ankylosed bone mass formation[J]. J Dent Res, 2015, 94(10): 1392-1400.
CHEN Y, KE J, LONG X, et al. Insulin-like growth factor-1 boosts the developing process of condylar hyperplasia by stimulating chondrocytes proliferation[J]. Osteoarthritis Cartilage, 2012, 20(4): 279-287.
CAO P, FENG Y, DENG M, et al. MiR-15b is a key regulator of proliferation and apoptosis of chondrocytes from patients with condylar hyperplasia by targeting IGF1, IGF1R and BCL2[J]. Osteoarthritis Cartilage, 2019, 27(2): 336-346.
WEN X, WANG Y X, GU Y. Transferrin promotes chondrogenic differentiation in condylar growth through inducing autophagy via ULK1-ATG16L1 axis[J]. Clin Sci, 2023, 137(18): 1431-1449.
BIOSSE DUPLAN M, KOMLA-EBRI D, HEUZÉ Y, et al. Meckel's and condylar cartilages anomalies in achondroplasia result in defective development and growth of the mandible[J]. Hum Mol Genet, 2016, 25(14): 2997-3010.
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突软骨下骨中的破骨细胞来源于骨髓中的单核-巨噬细胞.在下颌髁突生长发育的过程中,需要同时进行软骨细胞的增殖和破骨细胞介导的钙化软骨隔膜吸收,以完成软骨内成骨的过程[41].在这个过程中,首先需要破骨细胞前体的迁移,随后前体细胞分化为成熟细胞,并被相应的细胞因子激活后才能正常地行使功能.YANG等[42]研究发现WNT5A/ROR2(Wnt family member 5A/receptor tyrosine kinase like orphan receptor 2)信号激活能够促进破骨细胞前体的迁移和分化,从而导致软骨下骨破骨细胞活性增加,骨小梁骨质减少.此外,外界的机械应力也能激活软骨下骨中破骨细胞分化.KUANG等[43]发现,在机械应力的刺激下软骨细胞表达如基质细胞衍生因子-1(stromal cell-derived factor-1,SDF-1)、核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)等促破骨细胞分化因子.TIAN等[44]研究显示,过大的机械应力能激活髁突软骨下骨区成骨细胞中的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),增加RANKL/OPG(osteoclastogenesis inhibitory factor)比值,从而促进破骨细胞的形成.FENG等[45]研究显示,大鼠颞下颌关节盘前移位导致髁突表面受到的应力增大,通过激活RANTES/CCR/AKT2(regulated upon activation normal T cell expressed and secreted/C-C chemokine receptor/AKT serine kinase 2)信号轴增加破骨细胞的形成,因此过大的应力会导致软骨下骨的骨丢失.HE等[46]发现颞下颌关节强直的发生与破骨细胞异常相关,在颞下颌关节强直患者的髁突中检测到破骨细胞数量明显减少,并且单核细胞向破骨细胞的分化受阻,导致软骨下骨的骨重塑平衡被破坏.TANG等[47]研究发现,缺氧诱导因子-1的α亚单位(hypoxia inducible factor 1α,HIF-1α)通过单磷酸腺苷激活的蛋白激酶(adenosine 5'- monophosphate-activated protein kinase,AMPK)信号通路维持破骨细胞对髁突钙化软骨基质的吸收作用,以及通过介导血管内皮生长因子(vascular endothelial growth factor,VEGF)依赖性血管生成这两方面作用保证软骨下骨的正常形成.在小鼠破骨细胞中敲除Hif-1α后,由于破骨细胞数量缺乏及钙化软骨基质吸收功能障碍,导致髁突出现严重畸形,包括髁突长度缩短和纤维软骨层结构紊乱.综上所述,髁突软骨下骨的正常生长发育同时依赖于破骨细胞前体的迁移、破骨细胞分化以及成熟破骨细胞正常的功能三方面因素,异常外界应力或髁突内微环境的改变可能通过影响破骨细胞破坏软骨下骨稳态,影响软骨下骨的正常生长发育. ...
1
... 髁突肥大被认为是软骨细胞增殖异常增加及凋亡减弱的结果,而髁突发育不良则与之相反.CHEN等[48]分离髁突肥大患者的髁突软骨细胞,发现与正常软骨细胞相比,IGF-1的表达水平明显升高,高水平IGF-1通过MAPK/ERK(mitogen activated protein kinase/extracellular signal-regulated kinase)信号通路促进软骨细胞的增殖,同时增加软骨基质的合成,诱导髁突肥大的发生.在该研究结果的基础上,CAO等[49]研究发现miR-15b在髁突肥大患者的髁突软骨细胞中的表达显著降低.IGF-1、IGF-1受体及抗凋亡因子BCL2作为miR-15b的直接作用靶点,因miR-15b的抑制作用降低而出现表达升高,导致软骨细胞增殖异常增加,凋亡减弱,促进髁突肥大的发生. ...
1
... 髁突肥大被认为是软骨细胞增殖异常增加及凋亡减弱的结果,而髁突发育不良则与之相反.CHEN等[48]分离髁突肥大患者的髁突软骨细胞,发现与正常软骨细胞相比,IGF-1的表达水平明显升高,高水平IGF-1通过MAPK/ERK(mitogen activated protein kinase/extracellular signal-regulated kinase)信号通路促进软骨细胞的增殖,同时增加软骨基质的合成,诱导髁突肥大的发生.在该研究结果的基础上,CAO等[49]研究发现miR-15b在髁突肥大患者的髁突软骨细胞中的表达显著降低.IGF-1、IGF-1受体及抗凋亡因子BCL2作为miR-15b的直接作用靶点,因miR-15b的抑制作用降低而出现表达升高,导致软骨细胞增殖异常增加,凋亡减弱,促进髁突肥大的发生. ...
1
... 髁突发育不良的发生与髁突肥大相反,为软骨细胞凋亡异常增加,而增殖分化受阻的结果.WEN等[50]发现转铁蛋白(transferrin,TF)能够保护软骨细胞免受缺氧诱导的细胞凋亡,并且通过ULK1/ATG16L1(Unc-51 like autophagy activating kinase 1/autophagy related 16 like 1)信号轴诱导自噬,促进软骨细胞的分化.髁突发育不良患者血清中检测到TF水平降低,导致软骨细胞凋亡增加、增殖与分化受阻,最终引发疾病.此外,BIOSSE DUPLAN等[51]研究显示,在侏儒症患者中常见的成纤维细胞生长因子受体3(fibroblast growth factor receptor 3,FGFR3)功能获得性突变引发的受体过度激活会导致胚胎在下颌形成过程中麦氏软骨和髁突软骨的软骨细胞增殖分化能力缺陷,从而引发下颌骨发育不全及髁突畸形的发生. ...
1
... 髁突发育不良的发生与髁突肥大相反,为软骨细胞凋亡异常增加,而增殖分化受阻的结果.WEN等[50]发现转铁蛋白(transferrin,TF)能够保护软骨细胞免受缺氧诱导的细胞凋亡,并且通过ULK1/ATG16L1(Unc-51 like autophagy activating kinase 1/autophagy related 16 like 1)信号轴诱导自噬,促进软骨细胞的分化.髁突发育不良患者血清中检测到TF水平降低,导致软骨细胞凋亡增加、增殖与分化受阻,最终引发疾病.此外,BIOSSE DUPLAN等[51]研究显示,在侏儒症患者中常见的成纤维细胞生长因子受体3(fibroblast growth factor receptor 3,FGFR3)功能获得性突变引发的受体过度激活会导致胚胎在下颌形成过程中麦氏软骨和髁突软骨的软骨细胞增殖分化能力缺陷,从而引发下颌骨发育不全及髁突畸形的发生. ...



{kind=link}
{kind=link}
{kind=link}
{kind=link}