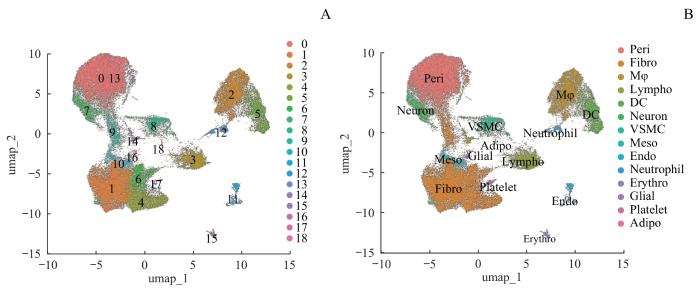
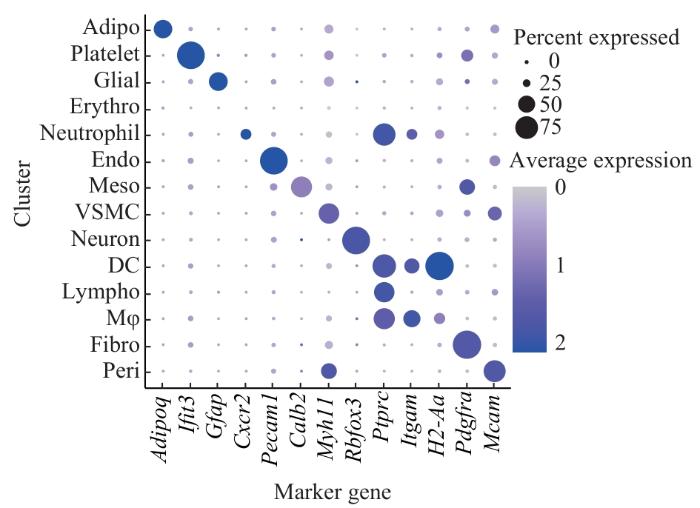
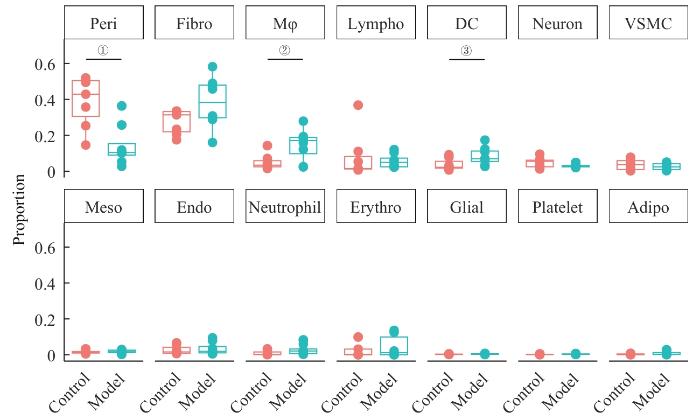
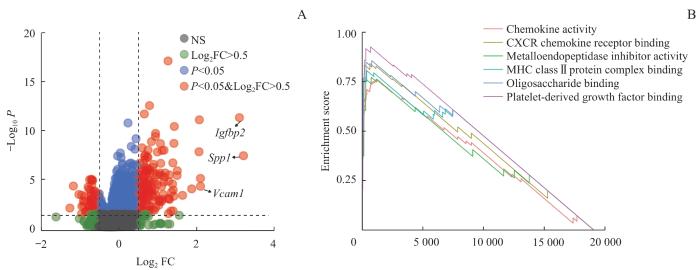
Objective ·To explore the single-cell landscape of aortic aneurysm (AA) utilizing single-cell RNA sequencing (scRNA-seq) technology. Methods ·A systematic search of the Gene Expression Omnibus (GEO) was conducted to collect all datasets meeting the inclusion criteria. Changes in the percentage of cellular composition of AA tissues versus normal control tissues were analyzed using R language and the Seurat package. Cell-cell interactions were assessed by gene expression levels of cellular receptor-ligand pairs using the CellChat package. Cellular senescence was scored and compared based on the SenMayo Senescence gene set using the AUCell package.Single-cell transcriptional data were simulated as traditional transcriptome data for differential gene screening and gene pathway enrichment analysis of pericytes. Results ·A total of nine datasets meeting the criteria were included. After quality control and merging, RNA count data for 104 570 cells were obtained, comprising 48 311 in the control group and 56 259 in the AA group. Cells were categorized into 19 clusters and annotated into 14 cell types. Compared with the control group, the proportion of pericytes in the AA group significantly decreased (P<0.001), while the proportions of monocytes/macrophages and dendritic cells increased (P=0.020, P=0.045). The number of intercellular interactions in the AA group was markedly higher than that in the control group; however, yet the interactions involving smooth muscle cells decreased, and the interaction intensity among pericytes diminished. There were 5 unique intercellular interactions in the control group and 13 unique interactions in the AA group, with the interaction involving SPP1 showing the highest relative information flow. Except for adipocytes, all cell types in the AA group exhibited significantly higher senescence scores (P<0.001), with an overall increase in the number of senescent cells (P<0.001), predominantly fibroblasts. Differential expression analysis of pericytes showed 185 upregulated genes and 151 downregulated genes in the AA group, with Spp1 exhibiting the highest upregulation. Pro-inflammatory pathways related to chemokine activity and CXC chemokine receptor binding were significantly enriched. Conclusion ·The cellular composition in AA tissues undergoes significant alterations, characterized by an increase in intercellular interactions and elevated levels of cellular senescence, with Spp1 identified as a key gene.
ZHANG Xingyu, LI Ruogu. Systematic analysis and exploration of single-cell transcriptomes in aortic aneurysm. Journal of Shanghai Jiao Tong University (Medical Science)[J], 2025, 45(6): 735-744 doi:10.3969/j.issn.1674-8115.2025.06.008
AA的scRNA-seq数据集均来自高通量基因表达数据库(Gene Expression Omnibus,GEO;http://www.ncbi.nlm.nih.gov/geo)。检索策略为("aneur*" OR "aneurism" OR "aneurysm") AND ("single cell" OR "singlecell" OR "single-cell" OR "scrna" OR "single neuc*" OR "single-neuc*" OR "snrna" OR "singleneuc*"),检索时间为2024年6月。数据集入选标准:①样本种属来源为小鼠。②对主动脉及 AA组织样本进行了scRNA-seq。③在同一项研究中同时包含 AA组及正常对照组。为保证各个数据集基因背景可比、细胞构成无偏倚,设定以下排除标准:①在进行测序前对样本细胞进行了特异性分选。②样本来源小鼠有其他与 AA建模无关的基因型背景。
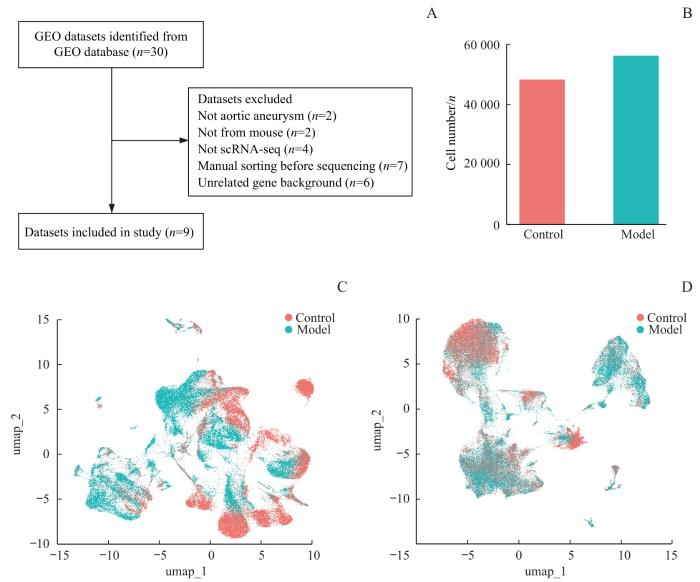
Note: A. Dataset screening flowchart. B. Cell numbers in each group. C/D. UMAP diagram of distribution of cells before (C) and after (D) Harmony integration.
Fig 1
Screening and integration of AA scRNA-seq datasets
Tab1
表1
表1各数据集的造模方法、取材时间及测序平台
Tab1 Induction method, harvest time point and sequencing platform of each dataset
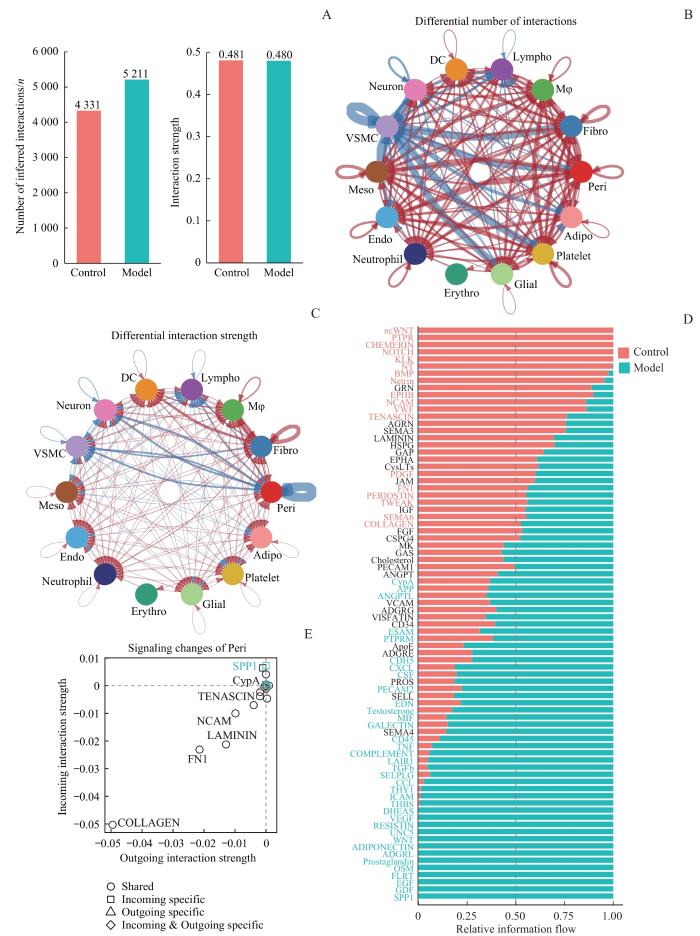
Note: A. Number and strength of inferred interactions in both groups. B. Differential number of interactions between the groups. Red arrows indicate a increased number of interactions in the AA group, and blue arrows indicate a decrease. C. Differential interaction strength between the groups. Red arrows indicate increased interaction strength in the AA group, and blue arrows indicate decreased strength. D. Overall information flow of each signaling pathway in both groups. E. Signaling changes of pericytes in the AA group compared to the control group.
Fig 5
Intergroup comparison of cell-cell interactions
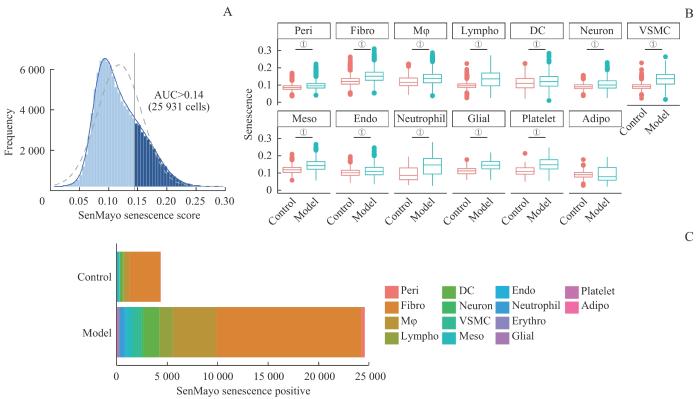
Note: A. Senescence-positive threshold determination based on the SenMayo senescence score. B. Comparison of SenMayo senescence scores for each cell type between the two groups. C. Number of senescence-positive cells of each cell type in both groups. ①P<0.001.
ZHANG Xingyu was responsible for research design, data acquisition and analysis, and paper writing. LI Ruogu was responsible for research topic selection, paper review, and revision. Both authors have read the last version of paper and agreed to the submission of the final manuscript.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
All authors disclose no relevant conflict of interests.
YANG H, DEROO E, ZHOU T, et al. Deciphering cell-cell communication in abdominal aortic aneurysm from single-cell RNA transcriptomic data[J]. Front Cardiovasc Med, 2022, 9: 831789.
BONTEKOE J, LIU B. Single-cell RNA sequencing provides novel insights to pathologic pathways in abdominal aortic aneurysm[J]. Front Cardiovasc Med, 2023, 10: 1172080.
WU H, XIE C, WANG R, et al. Comparative analysis of thoracic and abdominal aortic aneurysms across the segment and species at the single-cell level[J]. Front Pharmacol, 2022, 13: 1095757.
MIRANDA A M A, JANBANDHU V, MAATZ H, et al. Single-cell transcriptomics for the assessment of cardiac disease[J]. Nat Rev Cardiol, 2023, 20(5): 289-308.
CHOU E L, CHAFFIN M, SIMONSON B, et al. Aortic cellular diversity and quantitative genome-wide association study trait prioritization through single-nuclear RNA sequencing of the aneurysmal human aorta[J]. Arterioscler Thromb Vasc Biol, 2022, 42(11): 1355-1374.
DAVIS F M, TSOI L C, MA F, et al. Single-cell transcriptomics reveals dynamic role of smooth muscle cells and enrichment of immune cell subsets in human abdominal aortic aneurysms[J]. Ann Surg, 2022, 276(3): 511-521.
LI Y, REN P, DAWSON A, et al. Single-cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue[J]. Circulation, 2020, 142(14): 1374-1388.
HAO Y, STUART T, KOWALSKI M H, et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis[J]. Nat Biotechnol, 2024, 42(2): 293-304.
HAFEMEISTER C, SATIJA R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression[J]. Genome Biol, 2019, 20(1): 296.
KORSUNSKY I, MILLARD N, FAN J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony[J]. Nat Methods, 2019, 16(12): 1289-1296.
ARAN D, LOONEY A P, LIU L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage[J]. Nat Immunol, 2019, 20(2): 163-172.
JIN S, PLIKUS M V, NIE Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics[J]. Nat Protoc, 2025, 20(1): 180-219.
SAUL D, KOSINSKY R L, ATKINSON E J, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues[J]. Nat Commun, 2022, 13(1): 4827.
MACKAY C D A, JADLI A S, FEDAK P W M, et al. Adventitial fibroblasts in aortic aneurysm: unraveling pathogenic contributions to vascular disease[J]. Diagnostics (Basel), 2022, 12(4): 871.
MEEKEL J P, MATTEI G, COSTACHE V S, et al. A multilayer micromechanical elastic modulus measuring method in ex vivo human aneurysmal abdominal aortas[J]. Acta Biomater, 2019, 96: 345-353.
WANG D, HAO X, JIA L, et al. Cellular senescence and abdominal aortic aneurysm: from pathogenesis to therapeutics[J]. Front Cardiovasc Med, 2022, 9: 999465.
TAO W, HONG Y, HE H, et al. microRNA-199a-5p aggravates angiotensin II-induced vascular smooth muscle cell senescence by targeting Sirtuin-1 in abdominal aortic aneurysm[J]. J Cell Mol Med, 2021, 25(13): 6056-6069.
TETI G, CHIARINI F, MAZZOTTI E, et al. Cellular senescence in vascular wall mesenchymal stromal cells, a possible contribution to the development of aortic aneurysm[J]. Mech Ageing Dev, 2021, 197: 111515.
ASHIZAWA N, GRAF K, DO Y S, et al. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction [J]. J Clin Invest, 1996, 98(10): 2218-2227.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}