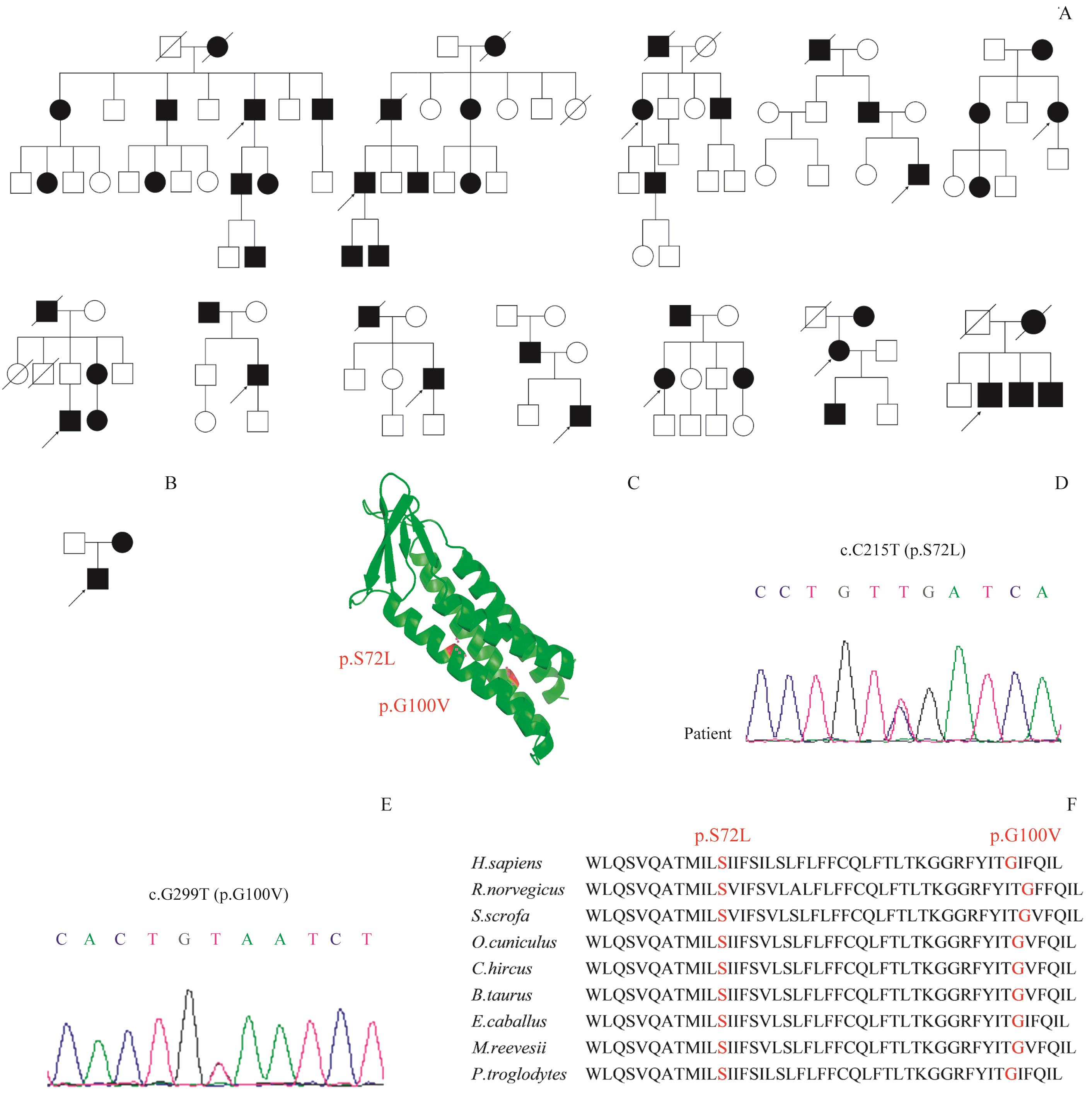
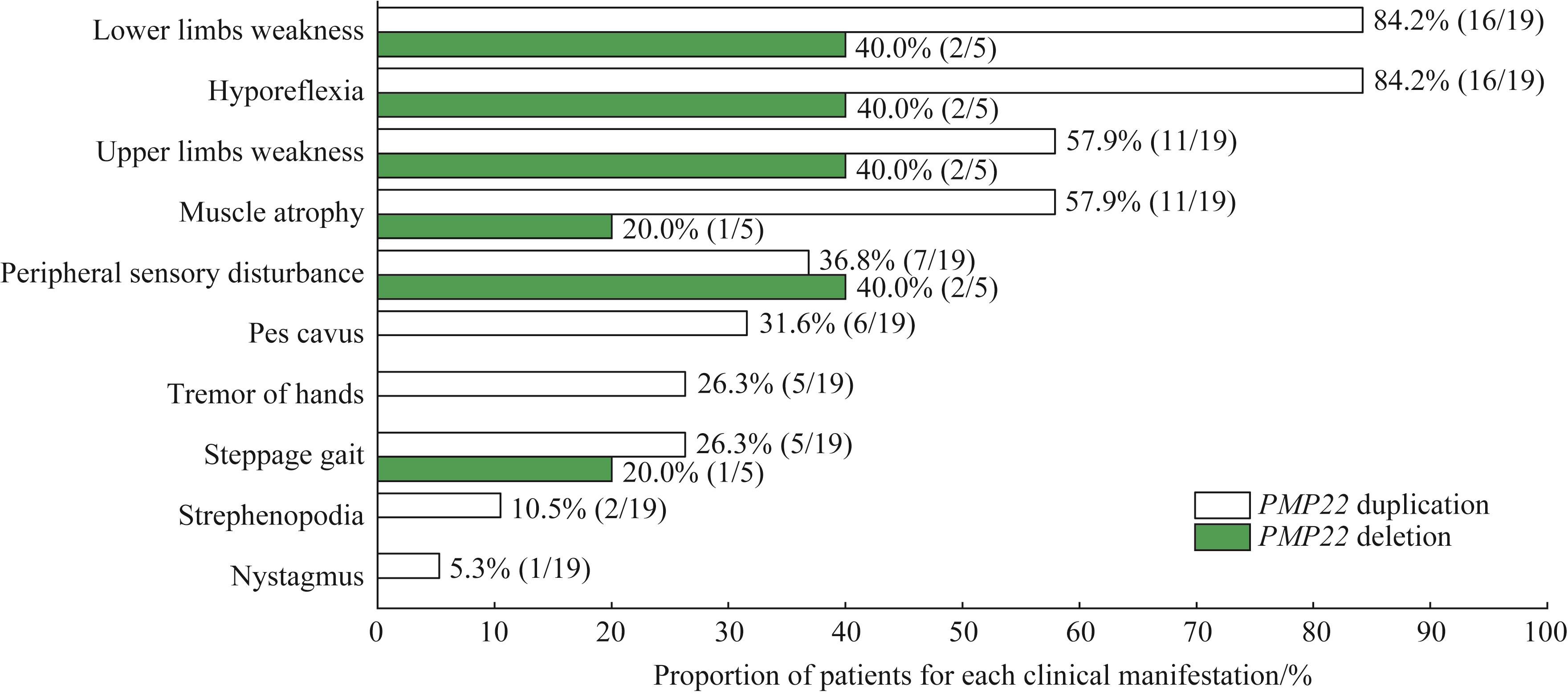
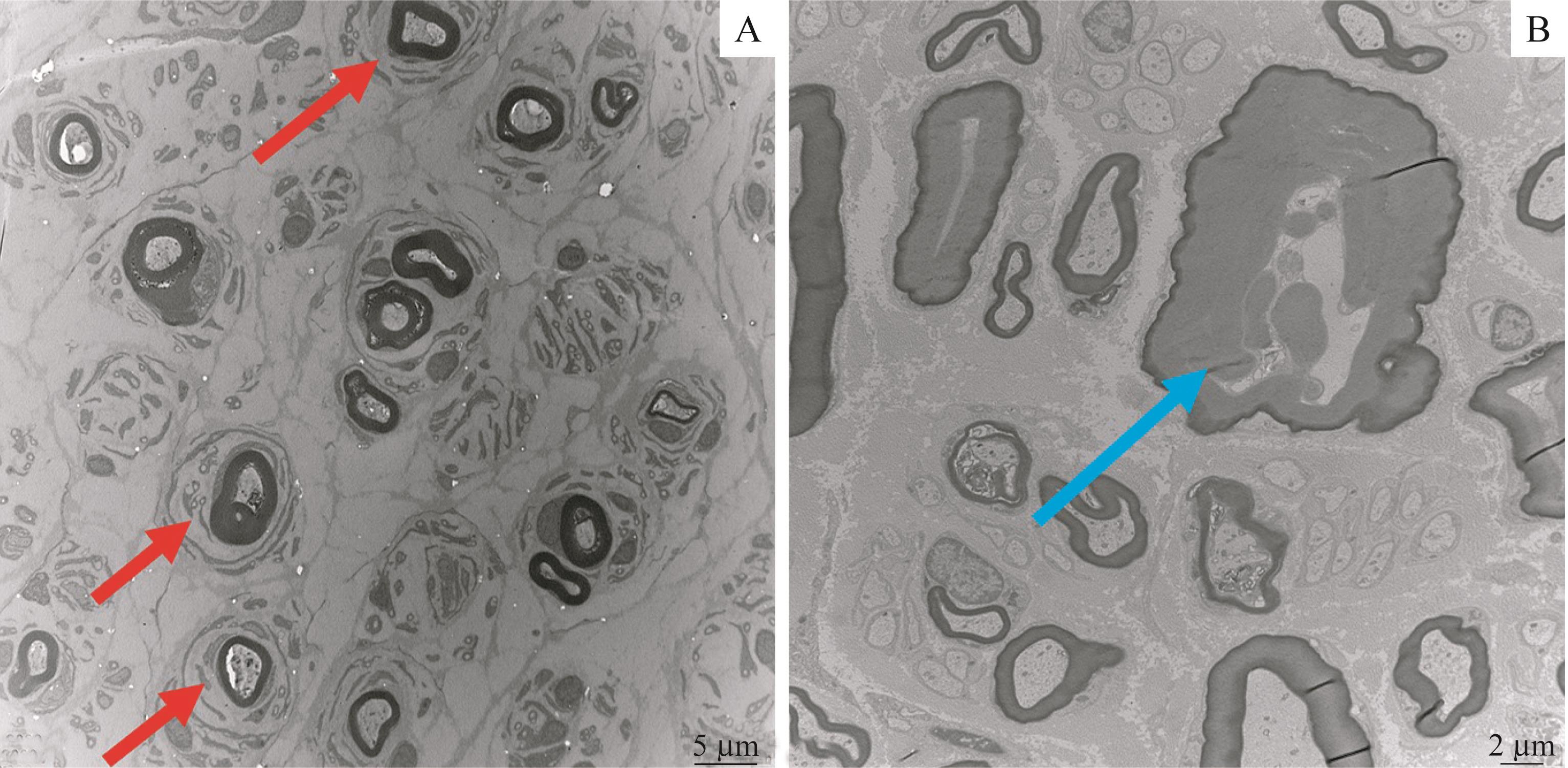
| 1 |
PANTERA H, MORAN J J, HUNG H A, et al. Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer[J]. Hum Mol Genet, 2018, 27(16): 2830-2839.
|
| 2 |
LIU X X, DUAN X H, ZHANG Y S, et al. Clinical and genetic diversity of PMP22 mutations in a large cohort of Chinese patients with Charcot-Marie-tooth disease[J]. Front Neurol, 2020, 11: 630.
|
| 3 |
朱啸巍, 钟平, 栾兴华. PMP22相关性周围神经病的临床及遗传学特点[J]. 中国实用神经疾病杂志, 2021, 24(14): 1265-1270.
|
|
ZHU X W, ZHONG P, LUAN X H. The clinical and genetic characteristics of PMP22-associated peripheral neuropathy[J]. Chin J Pract Nerv Dis, 2021, 24(14): 1265-1270.
|
| 4 |
GENTILE L, RUSSO M, FABRIZI G M, et al. Charcot-Marie-Tooth disease: experience from a large Italian tertiary neuromuscular center[J]. Neurol Sci, 2020, 41(5): 1239-1243.
|
| 5 |
WATILA M M, BALARABE S A. Molecular and clinical features of inherited neuropathies due to PMP22 duplication[J]. J Neurol Sci, 2015, 355(1/2): 18-24.
|
| 6 |
IVANOVIC V, BRANKOVIC M, BJELICA B, et al. Yield of the PMP22 deletion analysis in patients with compression neuropathies[J]. J Neurol, 2020, 267(12): 3617-3623.
|
| 7 |
D'ARRIGO S, TESSAROLLO V, TARONI F, et al. A case of severe early-onset neuropathy caused by a compound heterozygous deletion of the PMP22 gene: clinical and neurographic aspects[J]. Neuropediatrics, 2020, 51(3): 173-177.
|
| 8 |
MORINI A, MALAGUTI M C, MARANGONI S, et al. Neuropathic tremor in chronic inflammatory demyelinating polyneuropathy: the acquired equivalent of the roussy-levy syndrome[J]. Mov Disord Clin Pract, 2015, 3(2): 173-175.
|
| 9 |
徐佳露, 章毅, 赵聪颖, 等. 13例早发型腓骨肌萎缩症的基因分型研究[J]. 中国当代儿科杂志, 2019, 21(7): 670-675.
|
|
XU J L, ZHANG Y, ZHAO C Y, et al. A genotyping study of 13 cases of early-onset Charcot-Marie-Tooth disease[J]. Chin J Contemp Pediat, 2019, 21(7): 670-675.
|
| 10 |
ROA B B, DYCK P J, MARKS H G, et al. Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene[J]. Nat Genet, 1993, 5(3): 269-273.
|
| 11 |
MARQUES W Jr, THOMAS P K, SWEENEY M G, et al. Dejerine-Sottas neuropathy and PMP22 point mutations: a new base pair substitution and a possible “hot spot” on Ser72[J]. Ann Neurol, 1998, 43(5): 680-683.
|
| 12 |
RUSSO M, LAURÁ M, POLKE J M, et al. Variable phenotypes are associated with PMP22 missense mutations[J]. Neuromuscul Disord, 2011, 21(2): 106-114.
|
| 13 |
RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
|
| 14 |
WU R, LV H, ZHANG W, et al. Clinical and pathological variation of Charcot-Marie-tooth 1A in a large Chinese cohort[J]. Biomed Res Int, 2017, 2017: 6481367.
|
| 15 |
LOUSA M, VÁZQUEZ-HUARTE-MENDICOA C, GUTIÉRREZ A J, et al. Genetic epidemiology, demographic, and clinical characteristics of Charcot-Marie-tooth disease in the island of Gran Canaria (Spain)[J]. J Peripher Nerv Syst, 2019, 24(1): 131-138.
|
| 16 |
BROŽKOVÁ D, MAZANEC R, RYCHLÝ Z, et al. Four novel point mutations in the PMP22 gene with phenotypes of HNPP and Dejerine-Sottas neuropathy[J]. Muscle Nerve, 2011, 44(5): 819-822.
|
| 17 |
AZEVEDO H, PUPE C, PEREIRA R, et al. Pain in Charcot-Marie-Tooth disease: an update[J]. Arq Neuropsiquiatr, 2018, 76(4): 273-276.
|
| 18 |
KAWARAI T, YAMAZAKI H, MIYAMOTO R, et al. PMP22-related disease: a novel splice site acceptor variant and intrafamilial phenotype variability[J]. Neuromuscul Disord, 2019, 29(6): 422-426.
|
| 19 |
MITTENDORF K F, MARINKO J T, HAMPTON C M, et al. Peripheral myelin protein 22 alters membrane architecture[J]. Sci Adv, 2017, 3(7): e1700220.
|
| 20 |
ZHOU Y, MILES J R, TAVORI H, et al. PMP22 regulates cholesterol trafficking and ABCA1-mediated cholesterol efflux[J]. J Neurosci, 2019, 39(27): 5404-5418.
|
| 21 |
SVAREN J, MORAN J J, WU X Y, et al. Schwann cell transcript biomarkers for hereditary neuropathy skin biopsies[J]. Ann Neurol, 2019, 85(6): 887-898.
|
| 22 |
PANTERA H, SHY M E, SVAREN J. Regulating PMP22 expression as a dosage sensitive neuropathy gene[J]. Brain Res, 2020, 1726: 146491.
|
| 23 |
LI J. Genetic factors for nerve susceptibility to injuries: lessons from PMP22 deficiency[J]. Neural Regen Res, 2014, 9(18): 1661-1664.
|
| 24 |
TYSON J, ELLIS D, FAIRBROTHER U, et al. Hereditary demyelinating neuropathy of infancy. A genetically complex syndrome[J]. Brain, 1997, 120 ( Pt 1): 47-63.
|
| 25 |
NELIS E, HAITES N, VAN BROECKHOVEN C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies[J]. Hum Mutat, 1999, 13(1): 11-28.
|
| 26 |
WU R, FU J, MENG L C, et al. Homozygous splice-site mutation c.78+5G>A in PMP22 causes congenital hypomyelinating neuropathy[J]. Neuropathology, 2019, 39(6): 441-446.
|
| 27 |
LIAO Y C, TSAI P C, LIN T S, et al. Clinical and molecular characterization of PMP22 point mutations in Taiwanese patients with inherited neuropathy[J]. Sci Rep, 2017, 7(1): 15363.
|
| 28 |
RAMCHANDREN S. Charcot-Marie-tooth disease and other genetic polyneuropathies[J]. Continuum (Minneap Minn), 2017, 23(5, Peripheral Nerve and Motor Neuron Disorders): 1360-1377.
|
| 29 |
NAGAPPA M, SHARMA S, GOVINDARAJ P, et al. PMP22 gene-associated neuropathies: phenotypic spectrum in a cohort from India[J]. J Mol Neurosci, 2020, 70(5): 778-789.
|
| 30 |
ABE A, NAKAMURA K, KATO M, et al. Compound heterozygous PMP22 deletion mutations causing severe Charcot-Marie-Tooth disease type 1[J]. J Hum Genet, 2010, 55(11): 771-773.
|
| 31 |
AGOSTONI E, GOBESSI S, BRANCOLINI C, et al. Identification and characterization of a new member of the gas3/PMP22 gene family in C. elegans[J]. Gene, 1999, 234(2): 267-274.
|
| 32 |
ABE K T, LINO A M M, HIRATA M T A, et al. A novel stop codon mutation in the PMP22 gene associated with a variable phenotype[J]. Neuromuscul Disord, 2004, 14(5): 313-320.
|
| 33 |
TRACY J A, DYCK P J, KLEIN C J, et al. Onion-bulb patterns predict acquired or inherited demyelinating polyneuropathy[J]. Muscle Nerve, 2019, 59(6): 665-670.
|
| 34 |
MOSS K R, BOPP T S, JOHNSON A E, et al. New evidence for secondary axonal degeneration in demyelinating neuropathies[J]. Neurosci Lett, 2021, 744: 135595.
|
| 35 |
ZHAO H T, DAMLE S, IKEDA-LEE K, et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models[J]. J Clin Invest, 2018, 128(1): 359-368.
|
 ), 詹飞霞1, 张超2, 刘时华2, 钟平2, 曹立1,2, 栾兴华1,2(
), 詹飞霞1, 张超2, 刘时华2, 钟平2, 曹立1,2, 栾兴华1,2(