| Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
2
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
3
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
... [6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
... CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
2
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
... CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
2
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
... [11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... General information, and clinical and biochemical characteristics of ACALD patients Tab 1| Items | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 |
|---|
| Age of onset/Years | 27 | 23 | 40 | 37 | 34 | 30 | 33 | 38 | | Disease duration/Months | 12 | 4 | 59 | 31 | 5 | 4 | 8 | 16 | | First symptoms | Memory and cognitive decline | Dizziness with occipital numbness, insomnia | Mental and behavioral abnormalities | Dysarthria and ataxia | Bad temper, personality change | Memory and cognitive decline | Memory decline | Bad temper, personality change | | MMSE/MoCA | 21/17 | ND | NA | ND | 21/16 | 14/9 | 23/16 | NA | | VLCFA | | | | | | | | | | C26(≤1.30 nmol/mL) | 5.71 | ND | 3.94 | ND | 3.77 | 3.03 | 3.28 | 3.38 | | C24/C22(≤1.39) | 1.89 | ND | 1.50 | ND | 1.46 | 1.77 | 1.76 | 1.95 | | C26/C22(≤0.023) | 0.132 | ND | 0.065 | ND | 0.077 | 0.068 | 0.070 | 0.073 | | Adrenal insufficiency | | | | | | | | | | Cortisol levels | ↓ | ND | ↓ | ↓ | N | N | ↓ | ↓ | | ACTH | ↑ | ND | ↑ | ↑ | N | ↑ | ↑ | ↑ | | Protein content in cerebrospinal fluid | 1.00 g/L | N | 0.47 g/L | 0.47 g/L | N | ND | ND | ND | | Mutation information | | | | | | | | | | Exon | 8 | 10 | 6 | 7 | 1 | 1 | 6 | 3 | | Nucleotide mutations | c.1817C>T | c.2135G>A | c.1559T>C | c.1750delC | c.323C>T | c.160_170delACGCAGGAGGC | c.1534G>A | c.1202G>A | | Amino acid variants | S606L | R712H | L520P | H584Tfs*52 | S108L | T54Lfs*137 | G512S | R401Q | | ACMG | Pathogenic | VUS | VUS | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic | | Reported phenotypes | CCALD[3],AMN[4],AO[5] | NB[6] | NB[6] | ‒ | CCALD[7],AO[8], AMN[9], Olivo-ponto-cerebellar[10] | ‒ | CCALD[11],AMN[12],ACALD[11],AO[13],Female[14] | CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... CCALD[7],AMN[15],ACALD[16],NB[6]AO[17], ...
1
... Female carrier[18] ...
2
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
... [19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
2
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
... [25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
3
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
... [27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
... [27]. ...
1
... ACALD是一种高致死性疾病,未经治疗的患者平均在发病后7.5年死亡,在疾病早期进行异基因造血干细胞移植是阻止和延缓患者病情进展唯一有效方法[28].然而对于晚期患者,即Loes评分大于10分、扩展残疾状况量表(Expanded Disability Status Scale,EDSS)评分≥6分[46]、广泛锥体束受累和晚期严重认知障碍的患者[47],移植并不能提高患者的生存率,早期诊断是挽救患者生命的关键. ...
2
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
... [29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
... ALD临床异质性大.在国外的报道中,CALD主要累及儿童,而ACALD则较罕见,占ALD的2%~5%[19].在国内,CCALD较国外报道更多,约占本病的80%[20],ACALD仅占3.3%或更少[3,21].相较于儿童患者早期突出的学习障碍和行为问题,以及迅速的病程进展,临床上ACALD患者早期症状隐匿且首发症状表现多样,病程进展差异性大.认知功能障碍和精神行为异常是ACALD最常见的早期表现形式,尤其是当病变位于额叶时,患者症状类似于抑郁症或精神疾病[22].在疾病的早期,精神行为异常可能是患者唯一的症状,而无神经系统阳性体征[23].头部外伤及手术是诱发和加重病情的外部因素[24-25],多发生在3~12个月后.本研究中患者P7发病12个月前有明确的头部外伤史,但与既往报道[25]不同,该患者脱髓鞘部位与脑挫伤部位不一致.此外,患者P2和P3在发病前遭受情感打击.目前,尚无精神创伤诱发ALD发病的报道.精神创伤是否为发病的触发因素及其具体机制,需要更多的研究证实.本研究中,患者的发病年龄为(32.75±5.80)岁.目前,国内外尚无ACALD患者发病年龄的统计.但与AMN患者发病年龄[(27.6±8.7)岁]比较,ACALD患者发病稍晚[26].早期部分患者症状不典型,随着病情的进展,多数患者会出现疾病的各种症状[19],可能出现锥体束征及中枢性视觉异常,部分患者会有癫痫发作.本研究中,6例患者除有脑部受累的症状外,还表现为步态异常,其中5例患者需拐杖或轮椅助行.此外,本研究中的1例为罕见的橄榄脑桥小脑型ALD,其特征是头颅MRI表现为累及小脑和脑干脱髓鞘病变,临床主要表现为小脑共济失调、步态异常和语言障碍.此类患者常伴有假性球麻痹的症状,病程进展差异性大,患者生存期为3年到30余年不等;疾病的晚期,患者多死于严重的肺部感染[27].本研究中,患者P4以构音不清、共济失调起病,头颅MRI检查表现为典型的橄榄脑桥小脑型ALD白质病变模式.发病2年后,患者因严重的共济失调需要依赖轮椅,并有发音障碍及饮水呛咳的症状,病程进展较快.目前,全球所报道的病例主要分布在韩国、日本等亚洲地区[27-29],约占ALD的8.4%.而在中国大陆和台湾地区,仅有数例小脑型病例报道[29-32].这可能因为国内对该疾病认识不足,且部分患者病灶隐匿,仅在死后尸检发现小脑及脑干轻度脱髓鞘改变,因此易误诊为多系统萎缩[27]. ...
1
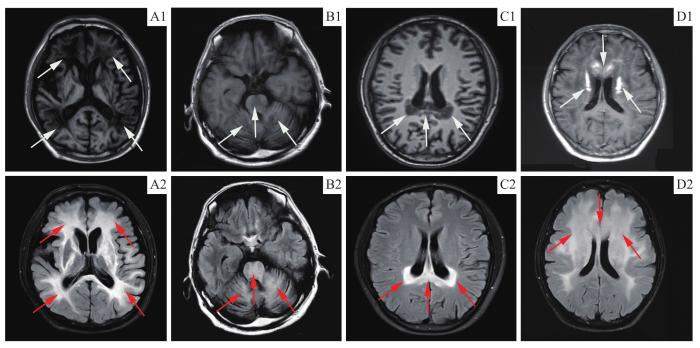
... 头颅MRI特征性病灶有助于疾病的诊断.在疾病早期,患者头颅MRI脱髓鞘病灶最常累及顶枕叶、胼胝体压部白质,双侧对称融合,呈蝶翼样外观,在T1序列呈低信号,在T2及液体衰减反转恢复(fluid attenuated inversion recovery,FLAIR)序列上呈高信号[33].少数患者最初表现为额顶叶及胼胝体膝部白质脱髓鞘的前部受累模式.随着病情的进展,在疾病晚期,白质广泛受累,脱髓鞘病灶呈弥漫性分布.本研究中,5例患者早期表现为典型的后部白质脱髓鞘模式.其中,P2就诊时处于疾病的早期,首发症状为持续性头晕、枕部麻木不适、失眠等非特异性改变,患者认知及行为正常,头颅MRI检查发现脑室旁多发白质脱髓鞘病灶,符合遗传代谢性脑白质病的影像学特征,最终经全外显子测序发现ABCD1基因突变而确诊为ALD.与既往报道一致[34],患者头颅MRI脱髓鞘改变多出现在明显的认知及行为异常之前.患者P3发病59个月时首次就诊于我院,此时已处于疾病的晚期,出现脑白质弥漫性脱髓鞘改变.此外,MRI增强检查发现病灶周围钆增强与活动性炎症脱髓鞘有关,是预后不良的表现[35].磁共振波谱成像是一种非侵入性的观察活体组织代谢变化的成像技术,通过不同的波峰及其峰值的大小反映组织内各化学物质的含量.其中,NAA仅表达于神经细胞中,其峰值的大小反映了神经元的功能状况;Cho是细胞膜磷脂代谢的成分之一,其峰值在脑恶性肿瘤及急性脱髓鞘疾病中升高[36].本研究,患者P1和P5的磁共振波谱成像检查均观察到了Cho峰升高及NAA峰减低,这说明在脑型CALD患者中,同时存在白质脱髓鞘和神经元的损伤. ...
1
... 头颅MRI特征性病灶有助于疾病的诊断.在疾病早期,患者头颅MRI脱髓鞘病灶最常累及顶枕叶、胼胝体压部白质,双侧对称融合,呈蝶翼样外观,在T1序列呈低信号,在T2及液体衰减反转恢复(fluid attenuated inversion recovery,FLAIR)序列上呈高信号[33].少数患者最初表现为额顶叶及胼胝体膝部白质脱髓鞘的前部受累模式.随着病情的进展,在疾病晚期,白质广泛受累,脱髓鞘病灶呈弥漫性分布.本研究中,5例患者早期表现为典型的后部白质脱髓鞘模式.其中,P2就诊时处于疾病的早期,首发症状为持续性头晕、枕部麻木不适、失眠等非特异性改变,患者认知及行为正常,头颅MRI检查发现脑室旁多发白质脱髓鞘病灶,符合遗传代谢性脑白质病的影像学特征,最终经全外显子测序发现ABCD1基因突变而确诊为ALD.与既往报道一致[34],患者头颅MRI脱髓鞘改变多出现在明显的认知及行为异常之前.患者P3发病59个月时首次就诊于我院,此时已处于疾病的晚期,出现脑白质弥漫性脱髓鞘改变.此外,MRI增强检查发现病灶周围钆增强与活动性炎症脱髓鞘有关,是预后不良的表现[35].磁共振波谱成像是一种非侵入性的观察活体组织代谢变化的成像技术,通过不同的波峰及其峰值的大小反映组织内各化学物质的含量.其中,NAA仅表达于神经细胞中,其峰值的大小反映了神经元的功能状况;Cho是细胞膜磷脂代谢的成分之一,其峰值在脑恶性肿瘤及急性脱髓鞘疾病中升高[36].本研究,患者P1和P5的磁共振波谱成像检查均观察到了Cho峰升高及NAA峰减低,这说明在脑型CALD患者中,同时存在白质脱髓鞘和神经元的损伤. ...
1
... 头颅MRI特征性病灶有助于疾病的诊断.在疾病早期,患者头颅MRI脱髓鞘病灶最常累及顶枕叶、胼胝体压部白质,双侧对称融合,呈蝶翼样外观,在T1序列呈低信号,在T2及液体衰减反转恢复(fluid attenuated inversion recovery,FLAIR)序列上呈高信号[33].少数患者最初表现为额顶叶及胼胝体膝部白质脱髓鞘的前部受累模式.随着病情的进展,在疾病晚期,白质广泛受累,脱髓鞘病灶呈弥漫性分布.本研究中,5例患者早期表现为典型的后部白质脱髓鞘模式.其中,P2就诊时处于疾病的早期,首发症状为持续性头晕、枕部麻木不适、失眠等非特异性改变,患者认知及行为正常,头颅MRI检查发现脑室旁多发白质脱髓鞘病灶,符合遗传代谢性脑白质病的影像学特征,最终经全外显子测序发现ABCD1基因突变而确诊为ALD.与既往报道一致[34],患者头颅MRI脱髓鞘改变多出现在明显的认知及行为异常之前.患者P3发病59个月时首次就诊于我院,此时已处于疾病的晚期,出现脑白质弥漫性脱髓鞘改变.此外,MRI增强检查发现病灶周围钆增强与活动性炎症脱髓鞘有关,是预后不良的表现[35].磁共振波谱成像是一种非侵入性的观察活体组织代谢变化的成像技术,通过不同的波峰及其峰值的大小反映组织内各化学物质的含量.其中,NAA仅表达于神经细胞中,其峰值的大小反映了神经元的功能状况;Cho是细胞膜磷脂代谢的成分之一,其峰值在脑恶性肿瘤及急性脱髓鞘疾病中升高[36].本研究,患者P1和P5的磁共振波谱成像检查均观察到了Cho峰升高及NAA峰减低,这说明在脑型CALD患者中,同时存在白质脱髓鞘和神经元的损伤. ...
1
... 头颅MRI特征性病灶有助于疾病的诊断.在疾病早期,患者头颅MRI脱髓鞘病灶最常累及顶枕叶、胼胝体压部白质,双侧对称融合,呈蝶翼样外观,在T1序列呈低信号,在T2及液体衰减反转恢复(fluid attenuated inversion recovery,FLAIR)序列上呈高信号[33].少数患者最初表现为额顶叶及胼胝体膝部白质脱髓鞘的前部受累模式.随着病情的进展,在疾病晚期,白质广泛受累,脱髓鞘病灶呈弥漫性分布.本研究中,5例患者早期表现为典型的后部白质脱髓鞘模式.其中,P2就诊时处于疾病的早期,首发症状为持续性头晕、枕部麻木不适、失眠等非特异性改变,患者认知及行为正常,头颅MRI检查发现脑室旁多发白质脱髓鞘病灶,符合遗传代谢性脑白质病的影像学特征,最终经全外显子测序发现ABCD1基因突变而确诊为ALD.与既往报道一致[34],患者头颅MRI脱髓鞘改变多出现在明显的认知及行为异常之前.患者P3发病59个月时首次就诊于我院,此时已处于疾病的晚期,出现脑白质弥漫性脱髓鞘改变.此外,MRI增强检查发现病灶周围钆增强与活动性炎症脱髓鞘有关,是预后不良的表现[35].磁共振波谱成像是一种非侵入性的观察活体组织代谢变化的成像技术,通过不同的波峰及其峰值的大小反映组织内各化学物质的含量.其中,NAA仅表达于神经细胞中,其峰值的大小反映了神经元的功能状况;Cho是细胞膜磷脂代谢的成分之一,其峰值在脑恶性肿瘤及急性脱髓鞘疾病中升高[36].本研究,患者P1和P5的磁共振波谱成像检查均观察到了Cho峰升高及NAA峰减低,这说明在脑型CALD患者中,同时存在白质脱髓鞘和神经元的损伤. ...
1
... VLCFA指由22个或者更多碳原子组成的脂肪酸.正常情况下,从食物中摄取和内源性合成的VLCFA通过β氧化的方式在过氧化物酶体内分解代谢;而在ALD患者中,由于β氧化功能障碍,使其聚积在血液及组织中.因此,几乎在所有的男性患者中均能发现血液中VLCFA水平的升高,尤其是C26及C26/C22不易受饮食及机体应激状态的影响[37],是筛查ALD的敏感指标.血清VLCFA升高是ALD特征性实验室指标,但VLCFA在ALD发病机制中的作用尚不明确.目前一些体内及体外研究表明,VLCFA可能通过诱导细胞产生氧化应激和炎症的方式导致组织损伤[38].本研究中6例进行检测的患者中,均发现血清VLCFA升高.但VLCFA水平与患者疾病的严重程度无关,且在不同的ALD类型中,也未发现VLCFA水平的差异[39].因此,VLCFA水平并不能用于监测疾病的进展.肾上腺皮质功能不全合并脑白质脱髓鞘是该病特征性表现,患者多表现为关节、乳头及牙龈等处的皮肤颜色变黑,厌食和体质量减轻[40].肾上腺皮质功能不全的终身患病率约为80%,约46.8%出现在小于10岁的儿童中.而在成人中,随着年龄的增长,患病率逐渐下降,仅约5.6%发生在40岁以上的患者中[39].鉴于肾上腺皮质功能不全的年龄相关性,对于小于40岁的成人患者,应每年检测一次肾上腺皮质功能;在40岁之后,如果出现内分泌系统症状,则可仅按需检测[39].本研究中,6例患者因肾上腺皮质功能不全需口服氢化可的松治疗.目前,尚无ALD患者何时进行激素替代治疗的统一标准.临床上多在患者出现促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)水平升高伴或不伴有皮质醇降低,或ACTH刺激试验异常(即皮质醇水平升高小于基线水平2倍)时开始口服激素替代治疗. ...
1
... VLCFA指由22个或者更多碳原子组成的脂肪酸.正常情况下,从食物中摄取和内源性合成的VLCFA通过β氧化的方式在过氧化物酶体内分解代谢;而在ALD患者中,由于β氧化功能障碍,使其聚积在血液及组织中.因此,几乎在所有的男性患者中均能发现血液中VLCFA水平的升高,尤其是C26及C26/C22不易受饮食及机体应激状态的影响[37],是筛查ALD的敏感指标.血清VLCFA升高是ALD特征性实验室指标,但VLCFA在ALD发病机制中的作用尚不明确.目前一些体内及体外研究表明,VLCFA可能通过诱导细胞产生氧化应激和炎症的方式导致组织损伤[38].本研究中6例进行检测的患者中,均发现血清VLCFA升高.但VLCFA水平与患者疾病的严重程度无关,且在不同的ALD类型中,也未发现VLCFA水平的差异[39].因此,VLCFA水平并不能用于监测疾病的进展.肾上腺皮质功能不全合并脑白质脱髓鞘是该病特征性表现,患者多表现为关节、乳头及牙龈等处的皮肤颜色变黑,厌食和体质量减轻[40].肾上腺皮质功能不全的终身患病率约为80%,约46.8%出现在小于10岁的儿童中.而在成人中,随着年龄的增长,患病率逐渐下降,仅约5.6%发生在40岁以上的患者中[39].鉴于肾上腺皮质功能不全的年龄相关性,对于小于40岁的成人患者,应每年检测一次肾上腺皮质功能;在40岁之后,如果出现内分泌系统症状,则可仅按需检测[39].本研究中,6例患者因肾上腺皮质功能不全需口服氢化可的松治疗.目前,尚无ALD患者何时进行激素替代治疗的统一标准.临床上多在患者出现促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)水平升高伴或不伴有皮质醇降低,或ACTH刺激试验异常(即皮质醇水平升高小于基线水平2倍)时开始口服激素替代治疗. ...
3
... VLCFA指由22个或者更多碳原子组成的脂肪酸.正常情况下,从食物中摄取和内源性合成的VLCFA通过β氧化的方式在过氧化物酶体内分解代谢;而在ALD患者中,由于β氧化功能障碍,使其聚积在血液及组织中.因此,几乎在所有的男性患者中均能发现血液中VLCFA水平的升高,尤其是C26及C26/C22不易受饮食及机体应激状态的影响[37],是筛查ALD的敏感指标.血清VLCFA升高是ALD特征性实验室指标,但VLCFA在ALD发病机制中的作用尚不明确.目前一些体内及体外研究表明,VLCFA可能通过诱导细胞产生氧化应激和炎症的方式导致组织损伤[38].本研究中6例进行检测的患者中,均发现血清VLCFA升高.但VLCFA水平与患者疾病的严重程度无关,且在不同的ALD类型中,也未发现VLCFA水平的差异[39].因此,VLCFA水平并不能用于监测疾病的进展.肾上腺皮质功能不全合并脑白质脱髓鞘是该病特征性表现,患者多表现为关节、乳头及牙龈等处的皮肤颜色变黑,厌食和体质量减轻[40].肾上腺皮质功能不全的终身患病率约为80%,约46.8%出现在小于10岁的儿童中.而在成人中,随着年龄的增长,患病率逐渐下降,仅约5.6%发生在40岁以上的患者中[39].鉴于肾上腺皮质功能不全的年龄相关性,对于小于40岁的成人患者,应每年检测一次肾上腺皮质功能;在40岁之后,如果出现内分泌系统症状,则可仅按需检测[39].本研究中,6例患者因肾上腺皮质功能不全需口服氢化可的松治疗.目前,尚无ALD患者何时进行激素替代治疗的统一标准.临床上多在患者出现促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)水平升高伴或不伴有皮质醇降低,或ACTH刺激试验异常(即皮质醇水平升高小于基线水平2倍)时开始口服激素替代治疗. ...
... [39].鉴于肾上腺皮质功能不全的年龄相关性,对于小于40岁的成人患者,应每年检测一次肾上腺皮质功能;在40岁之后,如果出现内分泌系统症状,则可仅按需检测[39].本研究中,6例患者因肾上腺皮质功能不全需口服氢化可的松治疗.目前,尚无ALD患者何时进行激素替代治疗的统一标准.临床上多在患者出现促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)水平升高伴或不伴有皮质醇降低,或ACTH刺激试验异常(即皮质醇水平升高小于基线水平2倍)时开始口服激素替代治疗. ...
... [39].本研究中,6例患者因肾上腺皮质功能不全需口服氢化可的松治疗.目前,尚无ALD患者何时进行激素替代治疗的统一标准.临床上多在患者出现促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)水平升高伴或不伴有皮质醇降低,或ACTH刺激试验异常(即皮质醇水平升高小于基线水平2倍)时开始口服激素替代治疗. ...
1
... VLCFA指由22个或者更多碳原子组成的脂肪酸.正常情况下,从食物中摄取和内源性合成的VLCFA通过β氧化的方式在过氧化物酶体内分解代谢;而在ALD患者中,由于β氧化功能障碍,使其聚积在血液及组织中.因此,几乎在所有的男性患者中均能发现血液中VLCFA水平的升高,尤其是C26及C26/C22不易受饮食及机体应激状态的影响[37],是筛查ALD的敏感指标.血清VLCFA升高是ALD特征性实验室指标,但VLCFA在ALD发病机制中的作用尚不明确.目前一些体内及体外研究表明,VLCFA可能通过诱导细胞产生氧化应激和炎症的方式导致组织损伤[38].本研究中6例进行检测的患者中,均发现血清VLCFA升高.但VLCFA水平与患者疾病的严重程度无关,且在不同的ALD类型中,也未发现VLCFA水平的差异[39].因此,VLCFA水平并不能用于监测疾病的进展.肾上腺皮质功能不全合并脑白质脱髓鞘是该病特征性表现,患者多表现为关节、乳头及牙龈等处的皮肤颜色变黑,厌食和体质量减轻[40].肾上腺皮质功能不全的终身患病率约为80%,约46.8%出现在小于10岁的儿童中.而在成人中,随着年龄的增长,患病率逐渐下降,仅约5.6%发生在40岁以上的患者中[39].鉴于肾上腺皮质功能不全的年龄相关性,对于小于40岁的成人患者,应每年检测一次肾上腺皮质功能;在40岁之后,如果出现内分泌系统症状,则可仅按需检测[39].本研究中,6例患者因肾上腺皮质功能不全需口服氢化可的松治疗.目前,尚无ALD患者何时进行激素替代治疗的统一标准.临床上多在患者出现促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)水平升高伴或不伴有皮质醇降低,或ACTH刺激试验异常(即皮质醇水平升高小于基线水平2倍)时开始口服激素替代治疗. ...
1
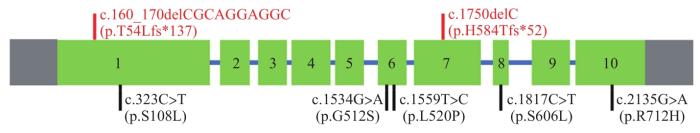
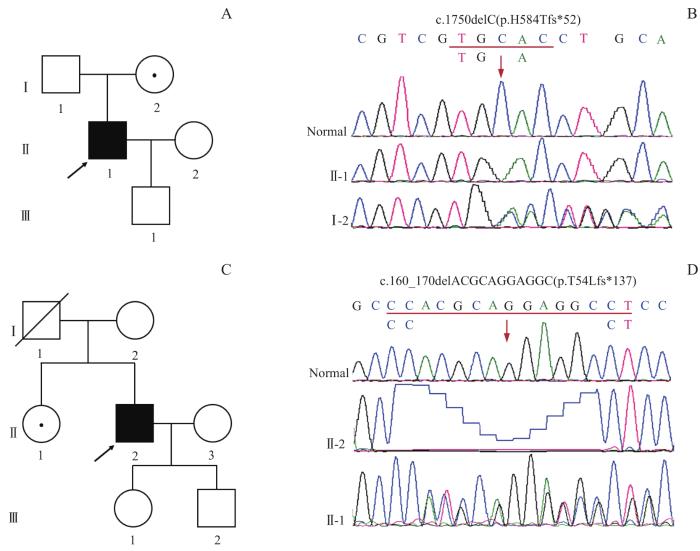
... ALD的发病率约为1∶14 700[41],ABCD1是唯一的致病基因,该基因由10个外显子和9个内含子组成.目前,有3 600余种ABCD1基因突变的报道(The ABCD1 Variant Database-Adrenoleukodystrophy.info).其中,错义突变是最常见的突变类型,约占50.4%;其次为移码突变,约占25.5%.1号外显子是国内外报道的热点突变外显子,约占所有突变的42.0%.本研究的病例中发现了8种不同的变异,其中6个为错义突变,2个为移码突变,与另一项国内报道相似[42],错义突变的比例均明显高于国外.此外,2个突变位于6号外显子,明显高于全球数据库中的11%,这一结果与之前国内及日本报道一致[43-44],即6号外显子是亚洲人群的另一个突变聚集区.然而,由于本研究中样本量少,这一结果尚需更多的数据进一步验证.本研究总结了6种已报道基因突变的表型分布特征,与既往的报道[45]一致,即患者的基因型和表型之间没有明显的相关性,即使在携带相同突变的同卵双生子中,患者也能出现不同的表型.2种突变(c.2135G>A和c.1559T>C)仅在新生儿筛查中发现,本文首次报道了其致病性表型.此外,本研究报道了ABCD1基因的2种新的移码突变(c.1750delC及c.160_170delACGCAGGAGGC),突变导致了ALDP翻译的提前终止而致病. ...
1
... ALD的发病率约为1∶14 700[41],ABCD1是唯一的致病基因,该基因由10个外显子和9个内含子组成.目前,有3 600余种ABCD1基因突变的报道(The ABCD1 Variant Database-Adrenoleukodystrophy.info).其中,错义突变是最常见的突变类型,约占50.4%;其次为移码突变,约占25.5%.1号外显子是国内外报道的热点突变外显子,约占所有突变的42.0%.本研究的病例中发现了8种不同的变异,其中6个为错义突变,2个为移码突变,与另一项国内报道相似[42],错义突变的比例均明显高于国外.此外,2个突变位于6号外显子,明显高于全球数据库中的11%,这一结果与之前国内及日本报道一致[43-44],即6号外显子是亚洲人群的另一个突变聚集区.然而,由于本研究中样本量少,这一结果尚需更多的数据进一步验证.本研究总结了6种已报道基因突变的表型分布特征,与既往的报道[45]一致,即患者的基因型和表型之间没有明显的相关性,即使在携带相同突变的同卵双生子中,患者也能出现不同的表型.2种突变(c.2135G>A和c.1559T>C)仅在新生儿筛查中发现,本文首次报道了其致病性表型.此外,本研究报道了ABCD1基因的2种新的移码突变(c.1750delC及c.160_170delACGCAGGAGGC),突变导致了ALDP翻译的提前终止而致病. ...
1
... ALD的发病率约为1∶14 700[41],ABCD1是唯一的致病基因,该基因由10个外显子和9个内含子组成.目前,有3 600余种ABCD1基因突变的报道(The ABCD1 Variant Database-Adrenoleukodystrophy.info).其中,错义突变是最常见的突变类型,约占50.4%;其次为移码突变,约占25.5%.1号外显子是国内外报道的热点突变外显子,约占所有突变的42.0%.本研究的病例中发现了8种不同的变异,其中6个为错义突变,2个为移码突变,与另一项国内报道相似[42],错义突变的比例均明显高于国外.此外,2个突变位于6号外显子,明显高于全球数据库中的11%,这一结果与之前国内及日本报道一致[43-44],即6号外显子是亚洲人群的另一个突变聚集区.然而,由于本研究中样本量少,这一结果尚需更多的数据进一步验证.本研究总结了6种已报道基因突变的表型分布特征,与既往的报道[45]一致,即患者的基因型和表型之间没有明显的相关性,即使在携带相同突变的同卵双生子中,患者也能出现不同的表型.2种突变(c.2135G>A和c.1559T>C)仅在新生儿筛查中发现,本文首次报道了其致病性表型.此外,本研究报道了ABCD1基因的2种新的移码突变(c.1750delC及c.160_170delACGCAGGAGGC),突变导致了ALDP翻译的提前终止而致病. ...
1
... ALD的发病率约为1∶14 700[41],ABCD1是唯一的致病基因,该基因由10个外显子和9个内含子组成.目前,有3 600余种ABCD1基因突变的报道(The ABCD1 Variant Database-Adrenoleukodystrophy.info).其中,错义突变是最常见的突变类型,约占50.4%;其次为移码突变,约占25.5%.1号外显子是国内外报道的热点突变外显子,约占所有突变的42.0%.本研究的病例中发现了8种不同的变异,其中6个为错义突变,2个为移码突变,与另一项国内报道相似[42],错义突变的比例均明显高于国外.此外,2个突变位于6号外显子,明显高于全球数据库中的11%,这一结果与之前国内及日本报道一致[43-44],即6号外显子是亚洲人群的另一个突变聚集区.然而,由于本研究中样本量少,这一结果尚需更多的数据进一步验证.本研究总结了6种已报道基因突变的表型分布特征,与既往的报道[45]一致,即患者的基因型和表型之间没有明显的相关性,即使在携带相同突变的同卵双生子中,患者也能出现不同的表型.2种突变(c.2135G>A和c.1559T>C)仅在新生儿筛查中发现,本文首次报道了其致病性表型.此外,本研究报道了ABCD1基因的2种新的移码突变(c.1750delC及c.160_170delACGCAGGAGGC),突变导致了ALDP翻译的提前终止而致病. ...
1
... ALD的发病率约为1∶14 700[41],ABCD1是唯一的致病基因,该基因由10个外显子和9个内含子组成.目前,有3 600余种ABCD1基因突变的报道(The ABCD1 Variant Database-Adrenoleukodystrophy.info).其中,错义突变是最常见的突变类型,约占50.4%;其次为移码突变,约占25.5%.1号外显子是国内外报道的热点突变外显子,约占所有突变的42.0%.本研究的病例中发现了8种不同的变异,其中6个为错义突变,2个为移码突变,与另一项国内报道相似[42],错义突变的比例均明显高于国外.此外,2个突变位于6号外显子,明显高于全球数据库中的11%,这一结果与之前国内及日本报道一致[43-44],即6号外显子是亚洲人群的另一个突变聚集区.然而,由于本研究中样本量少,这一结果尚需更多的数据进一步验证.本研究总结了6种已报道基因突变的表型分布特征,与既往的报道[45]一致,即患者的基因型和表型之间没有明显的相关性,即使在携带相同突变的同卵双生子中,患者也能出现不同的表型.2种突变(c.2135G>A和c.1559T>C)仅在新生儿筛查中发现,本文首次报道了其致病性表型.此外,本研究报道了ABCD1基因的2种新的移码突变(c.1750delC及c.160_170delACGCAGGAGGC),突变导致了ALDP翻译的提前终止而致病. ...
1
... ACALD是一种高致死性疾病,未经治疗的患者平均在发病后7.5年死亡,在疾病早期进行异基因造血干细胞移植是阻止和延缓患者病情进展唯一有效方法[28].然而对于晚期患者,即Loes评分大于10分、扩展残疾状况量表(Expanded Disability Status Scale,EDSS)评分≥6分[46]、广泛锥体束受累和晚期严重认知障碍的患者[47],移植并不能提高患者的生存率,早期诊断是挽救患者生命的关键. ...
1
... ACALD是一种高致死性疾病,未经治疗的患者平均在发病后7.5年死亡,在疾病早期进行异基因造血干细胞移植是阻止和延缓患者病情进展唯一有效方法[28].然而对于晚期患者,即Loes评分大于10分、扩展残疾状况量表(Expanded Disability Status Scale,EDSS)评分≥6分[46]、广泛锥体束受累和晚期严重认知障碍的患者[47],移植并不能提高患者的生存率,早期诊断是挽救患者生命的关键. ...
/
|
〈
|
![]()
|
〉
|
|
|
|
|
|
|
|
|
|
|
|
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}