

痤疮的病因和发病机制目前仍不清楚。越来越多的研究表明,微生物群失衡在痤疮的致病中起到至关重要的作用[4]。目前一些对痤疮微生态的研究对比了痤疮患者与健康人群皮肤表面菌群的差异:一项研究[5]表明,痤疮组与健康组皮肤微生物多样性无明显差异,假单胞菌、雷氏菌和假丝酵母以及念珠菌丰度发生明显改变;而另一项研究[6]提示痤疮患者皮肤表面微生物多样性大于健康对照,乳球菌属相对丰度增加。不同测序结果的差异可能与取材地点的气候、时间以及取材部位等多种因素有关。与皮肤表面不同,毛囊皮脂腺单位是一个相对密闭、缺氧且富含脂质的环境,其中的微生态结构与皮肤表面有所差异,并且受外界环境因素的影响较小,似乎与痤疮的发生更加密切。一项来自中国成都的观察性研究[7]比较了痤疮患者非炎性皮损和炎性皮损的菌群组成和差异,结果显示2组毛囊微生物α多样性差异无统计学意义。
尽管有关痤疮微生态的研究已有不少,但关于痤疮与微生物的关系仍存在许多未知。在痤疮发生的最早期阶段,毛囊漏斗部角化过度并引起毛囊皮脂腺导管堵塞,形成肉眼无法观察到的微粉刺。实际上,健康人群皮肤表面也可出现微粉刺,但为何只有痤疮患者会进一步发展出现后续丘疹、脓疱等皮损?此外,重度痤疮患者的皮损中存在着毛囊壁的破裂,当炎症进一步扩大时,导致结节、囊肿甚至后续瘢痕的形成,而中度痤疮患者皮损中则较少出现这样的情况,是何因素造成了痤疮皮损不同的发展结局?目前尚未有研究关注。因此,本研究拟通过16S rRNA高通量测序以及实时荧光定量聚合酶链反应(quantitative real-time polymerase chain reaction,qPCR)分析,阐明中度、重度痤疮患者非皮损区毛囊(即微粉刺)中的微生态差异。
1 对象与方法
1.1 研究对象
本研究为横断面研究,选取2022年8月—2023年8月在上海交通大学医学院附属仁济医院皮肤科就诊的中度、重度痤疮患者和健康志愿者。痤疮组纳入标准:① 年龄18~30岁。② 3个月内未使用过任何抗痤疮药物、物理治疗或医学美容治疗,未使用过口服或外用抗生素。③ Pillsbury分级[8]为2~3级的中度痤疮患者或Pillsbury分级为4级的重度痤疮患者。健康对照组为18~30岁健康体检者。研究对象的排除标准:① 近3个月使用过任何抗痤疮药物、物理治疗或医学美容治疗,以及口服或外用抗生素。② 存在其他面部疾病;有严重的心脑血管、肝、肾、自身免疫、肿瘤、血液、内分泌、神经、精神疾病和/或免疫缺陷。③ 参加其他临床试验研究者。
1.2 研究方法
1.2.1 标本收集
受试者清水洁面后端坐于治疗床上,操作者佩戴口罩、帽子、手套,70%的乙醇消毒整个鼻头3次,鼻头贴(花王,日本)粘贴于鼻部20 min后缓慢揭下,用高压灭菌后的镊子在立体显微镜下挑取30个毛囊内容物,均匀混悬于200 μL灭菌生理氯化钠溶液(上海上药信谊药厂有限公司)中,并放置于-80℃冰箱保存。
1.2.2 DNA提取和高通量测序
使用细菌基因组提取试剂盒[天根生化科技(北京)有限公司]提取全基因组DNA。用于PCR扩增的引物序列如表1所示,由上海华大基因科技有限公司合成。扩增产物在Illumina MiSeq平台PE300(因美纳,美国)上测序。
表1 引物序列
Tab 1
| Primer name | Primer sequence (5′→3′) | Amplified region |
|---|---|---|
| 341F | CCTACGGGNGGCWGCAG | V3‒4[9] |
| 805R | GACTACHVGGGTATCTAATCC |
1.2.3 qPCR
表2 qPCR绝对定量程序
Tab 2
| Step | Program |
|---|---|
| Step 1 | 95 ℃ 30 s |
| Step 2 (40 cycles) | 95 ℃ 5 s; 55 ℃ 30 s; 72 ℃ 30 s |
| Step 3 (dissociation stage) | 94 ℃ 30 s; 70 ℃ 1.5 min; 94 ℃ 10 s |
1.3 统计学分析
使用GraphPad Prism 9.0软件进行统计学分析和作图。定性资料以频数(百分率)表示,多组比较采用χ2检验。定量资料采用x±s表示;符合正态分布的定量资料,采用独立样本t检验进行组间比较;不符合正态分布的定量资料,采用Mann-Whitney U检验进行组间比较;采用Kruskal-Wallis检验进行多组数据比较。对β多样性进行基于bray-curtis距离的主坐标分析(principal coordinates analysis,PCoA)和Anosim相似性分析。将线性判别分析(linear discriminant analysis,LDA)值设为2时,进行LEfSe(LDA effect size)分析。P<0.05表示差异具有统计学意义。
2 结果
2.1 一般资料
共纳入10例中度痤疮患者(M组)、11例重度痤疮患者(S组)和11例健康志愿者(C组)。一般资料见表3,差异无统计学意义。
表3 3组人群一般资料
Tab 3
| Characteristic | C group (n=11) | M group (n=10) | S group (n=11) | H/χ² value | P value |
|---|---|---|---|---|---|
| Gender/n(%) | 0.182 | 0.913 | |||
| Male | 5 (45.5) | 5 (50.0) | 6 (54.5) | ||
| Female | 6 (54.5) | 5 (50.0) | 5 (45.5) | ||
| Age/year | 24.64±1.63 | 21.90±2.88 | 23.18±5.42 | 3.827 | 0.252 |
| Ethnic group/n(%) | 1.971 | 0.373 | |||
| Han | 10 (90.9) | 10 (100.0) | 11 (100.0) | ||
| Bai | 1 (9.1) | 0 (0) | 0 (0) | ||
| Education/n(%) | 2.676 | 0.262 | |||
| Senior high school/technical secondary school | 4 (36.4) | 2 (20.0) | 6 (54.5) | ||
| Junior college and above | 7 (63.6) | 8 (80.0) | 5 (45.5) | ||
| Work/n(%) | 1.703 | 0.427 | |||
| In service | 3 (27.3) | 2 (20.0) | 5 (45.5) | ||
| Not in service | 8 (72.7) | 8 (80.0) | 6 (54.5) | ||
| Habitual residence/n(%) | 2.147 | 0.342 | |||
| Shanghai | 11 (100.0) | 9 (90.0) | 9 (81.8) | ||
| Others | 0 (0) | 1 (10.0) | 2 (18.2) |
2.2 细菌V3~4区域高通量测序结果
2.2.1 α多样性分析
本研究中随着测序样本量的增加,物种累积曲线(图1A)趋于平缓,说明此环境中的物种并不会随样本量的增加而显著增多,表明本研究抽样充分。如图1B所示,C组、M组、S组菌群微生物操作分类单元(operational taxonomic unit,OTU)数分别有773、573、605个,其中361个OTU在3组中相同;C组和M组菌群有461个OTU相同,C组和S组菌群有437个OTU相同,M组和S组菌群有399个OTU相同。如图1C所示,C、M、S 3组样本中代表α多样性的Chao 1指数分别为301.43±52.82、208.55±85.19、247.28±30.04,C组与M组之间差异有统计学意义(P=0.020),即M组α多样性指数相对C组显著下降;S组相对于C组α多样性同样显著下降(P=0.013);而M与S组之间差异无统计学意义(P=0.314)。
图1
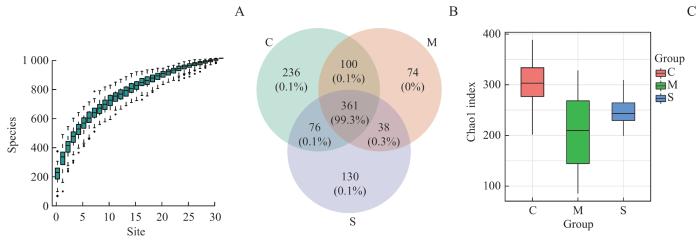
图1
3组人群毛囊菌群的α多样性分析
Note: A. Species accumulation curve. B. Venn diagrams of the OTU numbers of the three groups. C. Box plots of Chao1 index of the three groups.
Fig 1
α diversity analysis of follicular microbiome of the three groups
2.2.2 β多样性分析
β多样性是以物种群落间的距离来评估样本间物种群落的差异程度。PCoA图(图2)表明C组人群样本分布较为集中,组内差异较小;而M及S组人群样本分布有一定的趋势但较为离散,组内之间存在较大差异;3组样本趋势分布差异明显,说明其微生物群落结构差异较大。相似性分析结果显示C组与M组(P=0.027)、C组与S组(P=0.017)组间β多样性差异有统计学意义,物种组成相似度低;M组与S组(P=0.160)组间物种相似度较高。
图2
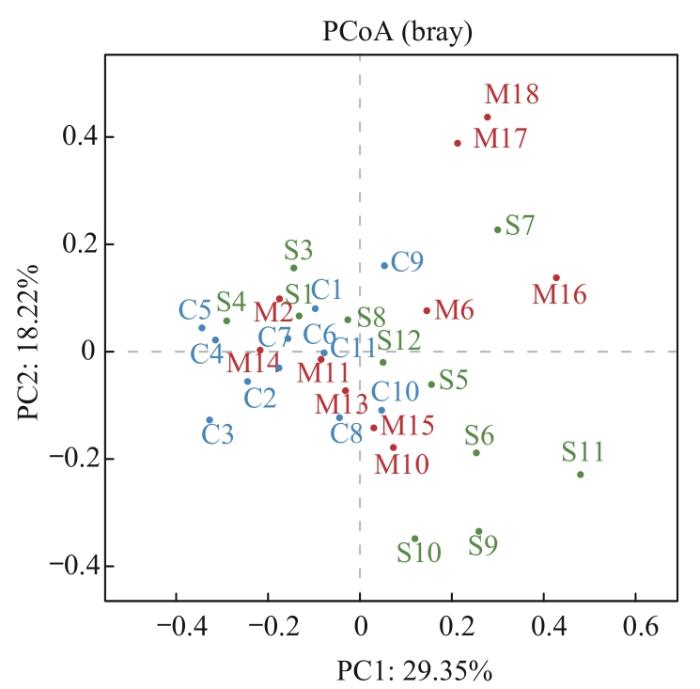
图2
3组人群毛囊菌群的β多样性分析
Note: PCoA scatter plot based on β diversity index between samples.
Fig 2
β-diversity analysis of hair follicle microbiome of the three groups
2.2.3 物种组成分析
按照门、属划分的3组人群细菌菌群的物种相对丰度见表4~5。如图3所示,门水平上3组的优势菌群均为放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)以及拟杆菌门(Bacteroidetes)。相对于C组,M组中变形菌门相对丰度显著降低(P=0.043),S组中厚壁菌门相对丰度显著升高(P=0.040)。如图4所示,属水平上C组菌群相对丰度排名前五的菌属依次为丙酸杆菌属(Propionibacterium)、未分类的放线菌属(unclassified Actinomycetales)、葡萄球菌属(Staphylococcus)、未分类的奈瑟菌属(unclassified Neisseriaceae)以及类芽孢杆菌属(Caldalkalibacillus);M组和S组菌群相对丰度排名前五的菌属均为葡萄球菌属、丙酸杆菌属、未分类的放线菌属、棒状杆菌属(Corynebacterium)以及未分类的奈瑟菌属。随着痤疮严重度增加,丙酸杆菌属相对丰度呈现下降趋势,但差异无统计学意义;相对于C组,M组和S组毛囊中的葡萄球菌属相对丰度均显著增加(P=0.010,P=0.019)。
表4 3组样本细菌菌群在门水平上的丰度(%)
Tab 4
| Phylum | Abundance/% | ||
|---|---|---|---|
| C group (n=11) | M group (n=10) | S group (n=11) | |
| Actinobacteria | 61.48±19.39 | 58.48±17.59 | 49.64±28.26 |
| Firmicutes | 21.94±16.10 | 35.42±18.37 | 43.90±26.42 |
| Proteobacteria | 14.84±15.23 | 5.27±5.79 | 5.51±5.41 |
| Bacteroidetes | 0.86±0.88 | 0.27±0.29 | 0.59±1.13 |
| Others | 0.88±0.55 | 0.56±0.57 | 0.36±0.49 |
表5 3组样本细菌菌群在属水平上的丰度(%)
Tab 5
| Genus | Abundance/% | ||
|---|---|---|---|
C group (n=11) | M group (n=10) | S group (n=11) | |
| Staphylococcus | 13.05±11.54 | 31.54±17.84 | 37.30±27.09 |
| Propionibacterium | 36.99±17.67 | 25.94±19.63 | 23.78±16.30 |
| Unclassified Actinomycetales | 18.78±19.20 | 23.38±15.96 | 19.58±17.88 |
| Corynebacterium | 2.09±2.77 | 8.28±11.28 | 5.47±4.74 |
| Unclassified Neisseriaceae | 4.36±8.64 | 0.06±0.08 | 0.86±2.48 |
| Others | 24.73±22.90 | 10.80±9.32 | 13.01±22.05 |
图3
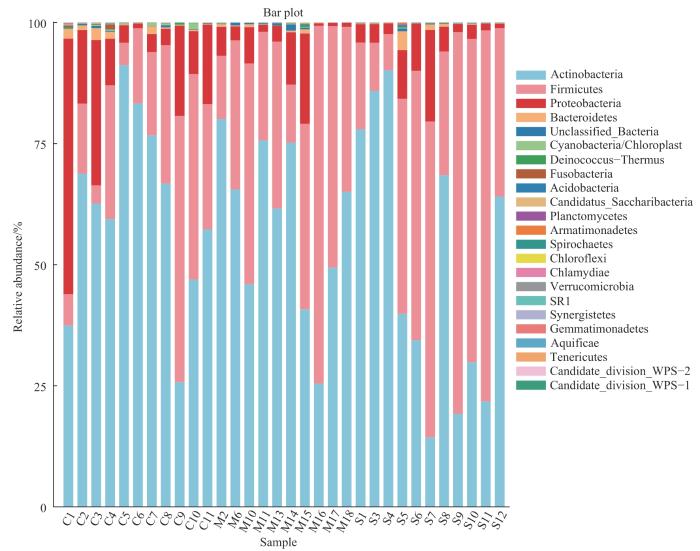
图3
3组人群毛囊菌群在门水平上相对丰度柱状图
Fig 3
Column diagram of relative abundance of hair follicle microbiome at the phylum level in the three groups
图4
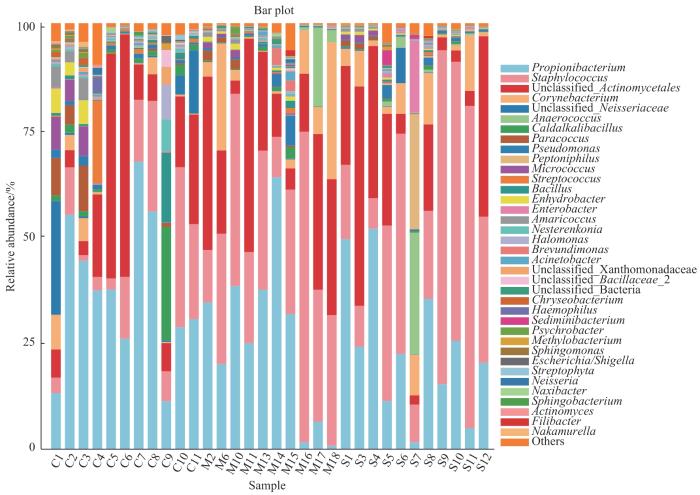
图4
3组人群毛囊菌群在属水平上相对丰度柱状图
Fig 4
Column diagram of relative abundance of hair follicle microbiome at the genus level in the three groups
LEfSe分析通过2组之间的比较,从而找到组间在丰度上有显著差异的特征或者生物标志物(物种),进一步显示了微生物组成的差异。如图5所示,与C组相比,葡萄球菌属(LDA=5.50)在M组中起重要作用。如图6所示,与C组相比,葡萄球菌属(LDA=5.57)、棒状杆菌属(LDA=4.74)、沉积杆菌属(Sediminibacterium,LDA=3.62)、Unclassified_Acidobacteria_Gp3(LDA=2.66)以及Gemmata(LDA=2.19)在S组中起重要作用。如图7所示,与M组相比,Unclassified_Acidobacteria_Gp3(LDA=2.66)在S组中起重要作用。
图5
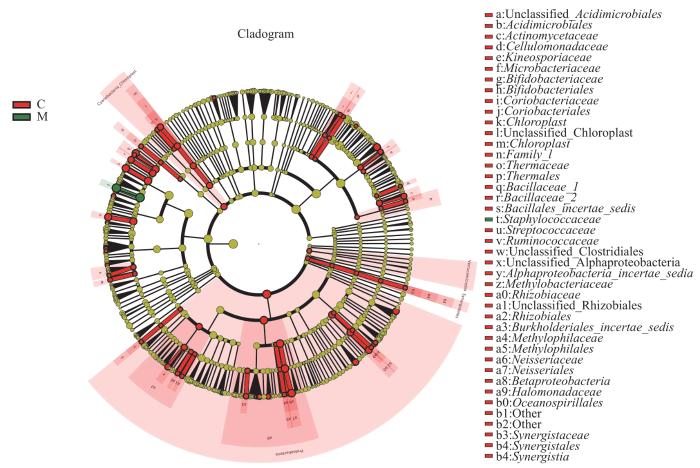
图5
C组和M组人群毛囊菌群LEfSe分析进化分支图
Note: The circles radiating from the inside out represent the classification levels from phylum to genus. Each circle and its shaded range at different classification levels represent the classification at that level, and the diameter of the circle is proportional to the relative abundance of the species. The species represented by the English letters next to the circles are shown in the legend. The red nodes represent microbial communities with high abundance and important roles in the red group, the green nodes represent microbial communities with important roles in the green group, and the yellow nodes represent species with no differences between groups.
Fig 5
LEfSe cladogram of analysis of follicular microbiome in C group and M group
图6
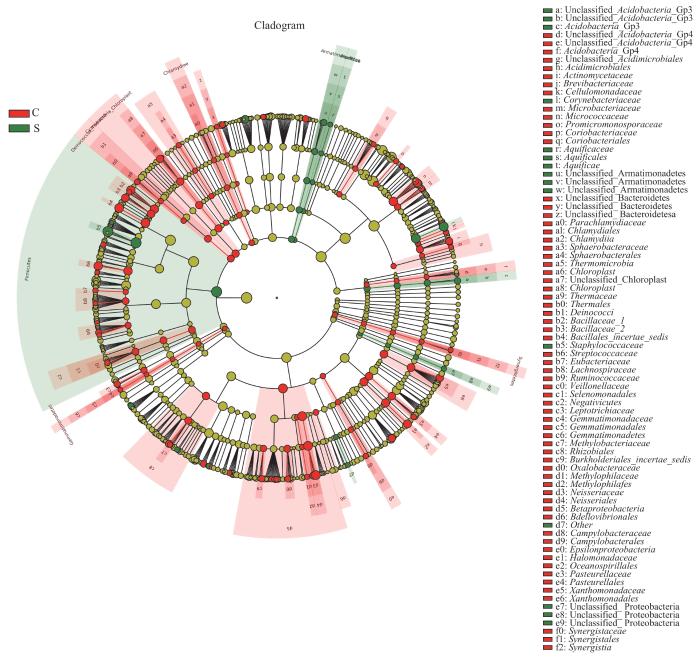
图6
C组和S组人群毛囊菌群LEfSe分析进化分支图
Note: The circles radiating from the inside out represent the classification levels from phylum to genus. Each circle and its shaded range at different classification levels represent the classifications at those levels, and the diameter of the circle is proportional to the relative abundance of the species. The species represented by the English letters next to the circles are shown in the legend. The red nodes represent microbial communities with high abundance and important roles in the red group, the green nodes represent microbial communities with important roles in the green group, and the yellow nodes represent species with no differences between groups.
Fig 6
LEfSe cladogram of analysis of hair follicle microbiome in Group C and Group S
图7
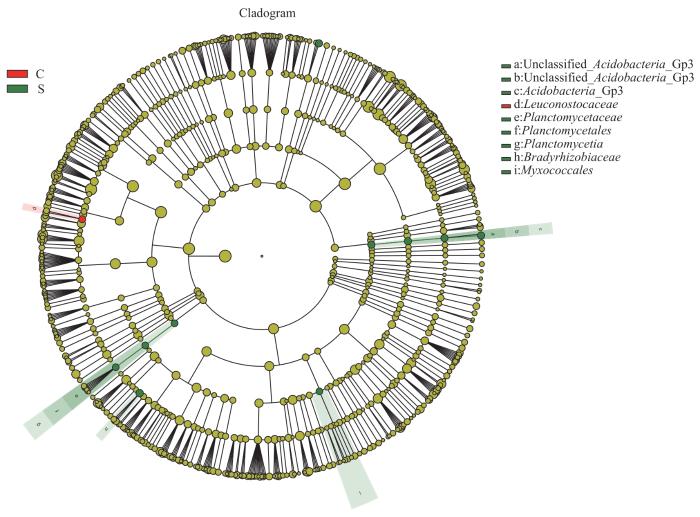
图7
M组和S组人群毛囊菌群LEfSe分析进化分支图
Note: The circles radiating from the inside out represent the classification level from phylum to genus. Each circle and its shaded range at different classification levels represent the classification at that level, and the diameter of the circle is proportional to the relative abundance of the species. The species represented by the English letters next to the circle are shown in the legend. The red nodes represent microbial communities with high abundance and important roles in the red group, the green nodes represent microbial communities with important roles in the green group, and the yellow nodes represent species with no differences between groups.
Fig 7
LEfSe cladogram of analysis of hair follicle microbiome in Group M and Group S
2.3 细菌绝对定量差异分析
qPCR结果如图8所示,C组细菌总拷贝数为(17 262.28±11 439.72)个/μL,M组和S组分别为(67 506.34±38 265.74)个/μL和(188 674.75±141 689.54)个/μL。相对于C组,M组和S组样本中细菌拷贝数均显著增加(均P=0.001);S组毛囊样本中细菌载量显著高于M组,差异有统计学意义(P=0.017)。
图8
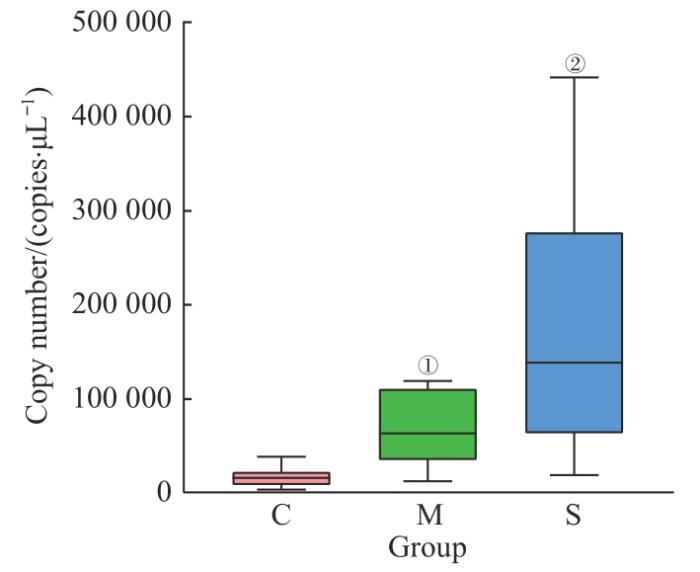
图8
3组人群毛囊细菌总拷贝数箱型图
Fig 8
Box plots of total number of follicular bacterial copies of the three groups
3 讨论
本研究为明确中度痤疮、重度痤疮以及健康对照人群组间非皮损区毛囊菌群结构差异,对不同组别样本的毛囊菌群进行了α及β多样性分析。α多样性分析发现,中度痤疮组和重度痤疮组相对于健康组菌群的Chao1指数差异有统计学意义,痤疮患者的毛囊菌群多样性较健康对照组明显降低,这一结论再次佐证了相对多样化的菌群是健康的微生物群这一观点[14]。基于Bray-Curtis距离的PCoA图表明健康组人群样本分布较为集中,组内差异较小;而中度痤疮组及重度痤疮组人群样本分布有一定的趋势但较为离散,组内之间存在较大差异;3组样本趋势分布差异明显,说明其微生物群落结构差异较大。相似性分析结果显示健康组与痤疮组相比β多样性有显著差异,物种组成相似度低;2个痤疮组组间物种相似度较高。
本研究进一步对3组人群毛囊菌群的群落结构进行分析,发现在门水平上3组样本的优势细菌主要是由放线菌门、厚壁菌门、变形菌门、拟杆菌门组成,占比大于99%。相对于健康组人群,中度痤疮组毛囊中变形菌门相对丰度显著降低,重度痤疮组厚壁菌门的相对丰度显著升高。这提示痤疮患者毛囊菌群的多样性与痤疮的严重程度可能存在某种相关性。属水平上健康组优势菌为丙酸杆菌属、未分类的放线菌属,2个痤疮组优势菌为葡萄球菌属、丙酸杆菌属。随着痤疮严重度增加,丙酸杆菌属相对丰度呈现下降趋势,但数据无统计学意义;相对于健康组,中度痤疮组和重度痤疮组非皮损区毛囊中的葡萄球菌属相对丰度均显著增加。
传统观点认为,痤疮的发生与痤疮丙酸杆菌(Cutibacterium acnes,C. acnes)的过度增殖有关,C. acnes能够分泌脂肪酶、透明质酸酶和蛋白酶,产生代谢物卟啉、短链脂肪酸等毒力因子,促进角质细胞增殖、诱导分化、炎症反应等诱发或加重痤疮的病情[15-16]。然而FITZ-GIBBON等[17]发现在健康者和痤疮患者的毛囊皮脂腺单位中,C. acnes均为最丰富的细菌,细菌丰度没有差异,而菌株种群结构显著不同。本研究进一步佐证了C. acnes并非痤疮发生的单一因素。此外,本研究通过LEfSe分析发现中度痤疮组和重度痤疮组中的葡萄球菌属均显著增加,提示其可能成为疾病发生的关键微生物。表皮葡萄球菌(Staphylococcus epidermidis,S. epidermidis)是毛囊皮脂腺单位中的另一代表性微生物,DRENO等[18]的研究表明在丘疹、脓疱皮损中S. epidermidis是最丰富的物种,但其在痤疮中具体作用及其机制尚不清晰。有研究发现其分泌的细菌素及代谢产物短链脂肪酸如乙酸、丁酸可以抑制C. acnes的定植和传播[19];其病原相关分子模式脂磷壁酸刺激角质形成细胞表面Toll样受体2(Toll-like receptor 2,TLR2)产生miRNA⁃143,后者可以降低TLR2分子的表达数量,从而减少C. acnes通过TLR2产生的促炎因子[20]。表皮素(epidermin)为S. epidermidis分泌的羊毛硫抗生素家族主要成员之一,在极低浓度下便可作用于多种革兰阳性杆菌,通过与靶细胞壁上的脂质Ⅱ结合使其穿孔导致细菌死亡[21]。S. epidermidis产生的电能还可以抑制C. acnes的生长以及造成细胞裂解[22]。另一方面,S. epidermidis可能作为致病菌参与痤疮的发生。DAGNELIE等[23]证实S. epidermidis似乎比C. acnes在诱导先天免疫分子尤其是白细胞介素-6(interleukin-6,IL-6)、γ干扰素诱导蛋白-10(interferon γ-inducible protein-10,IP-10)等方面具有更强的作用。
综上所述,本研究结合16S rRNA测序以及qPCR探索了不同严重程度痤疮患者非皮损区毛囊菌群多样性、物种组成和载量的差异,表明在痤疮患者的最早期皮损(微粉刺)中微生态已经发生了改变。痤疮的发生及严重程度可能与毛囊菌群变化存在着密切的联系。这一发现可能为深入探究微生物介导的痤疮恶化机制,以及开发基于微生物的痤疮治疗方法提供有力的证据。基于本研究中筛选出的特异性菌群标志物,后期将扩大样本量、结合多组学分析和体内动物实验等进一步明确其是否参与疾病发生发展及相关作用机制。
作者贡献声明
梁梦晨负责研究的构思、设计和实施;梁梦晨、李嘉祺、莫小辉负责论文撰写与修改;梁梦晨、李嘉祺负责菌群检测和统计分析;梁梦晨、吴心怡负责生物样本的采集;鞠强参与论文审阅与修改。所有作者均阅读并同意了最终稿件的提交。
AUTHOR's CONTRIBUTIONS
The study was conceived, designed, and implemented by LIANG Mengchen. The manuscript was drafted and revised by LIANG Mengchen, LI Jiaqi and MO Xiaohui. The microbiota analysis and statistical analysis were performed by LIANG Mengchen and LI Jiaqi. The biosamples were collected by LIANG Mengchen and WU Xinyi. JU Qiang reviewed and revised the manuscript. All the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
All authors disclose no relevant conflict of interests.
参考文献

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}