JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE) ›› 2021, Vol. 41 ›› Issue (5): 571-578.doi: 10.3969/j.issn.1674-8115.2021.05.003
• Basic research • Previous Articles Next Articles
Lu-di YANG1( ), Gao-ming WANG1, Ren-hao HU2, Xiao-hua JIANG2, Ran CUI2()
), Gao-ming WANG1, Ren-hao HU2, Xiao-hua JIANG2, Ran CUI2()
Online:2021-05-28
Published:2021-05-27
Contact:
Ran CUI
E-mail:yangludi@sjtu.edu.cn;cuiangus@tongji.edu.cn
Supported by:CLC Number:
Lu-di YANG, Gao-ming WANG, Ren-hao HU, Xiao-hua JIANG, Ran CUI. Identification of core genes in pancreatic cancer progression by bioinformatics analysis[J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(5): 571-578.
Add to citation manager EndNote|Ris|BibTeX
URL: https://xuebao.shsmu.edu.cn/EN/10.3969/j.issn.1674-8115.2021.05.003
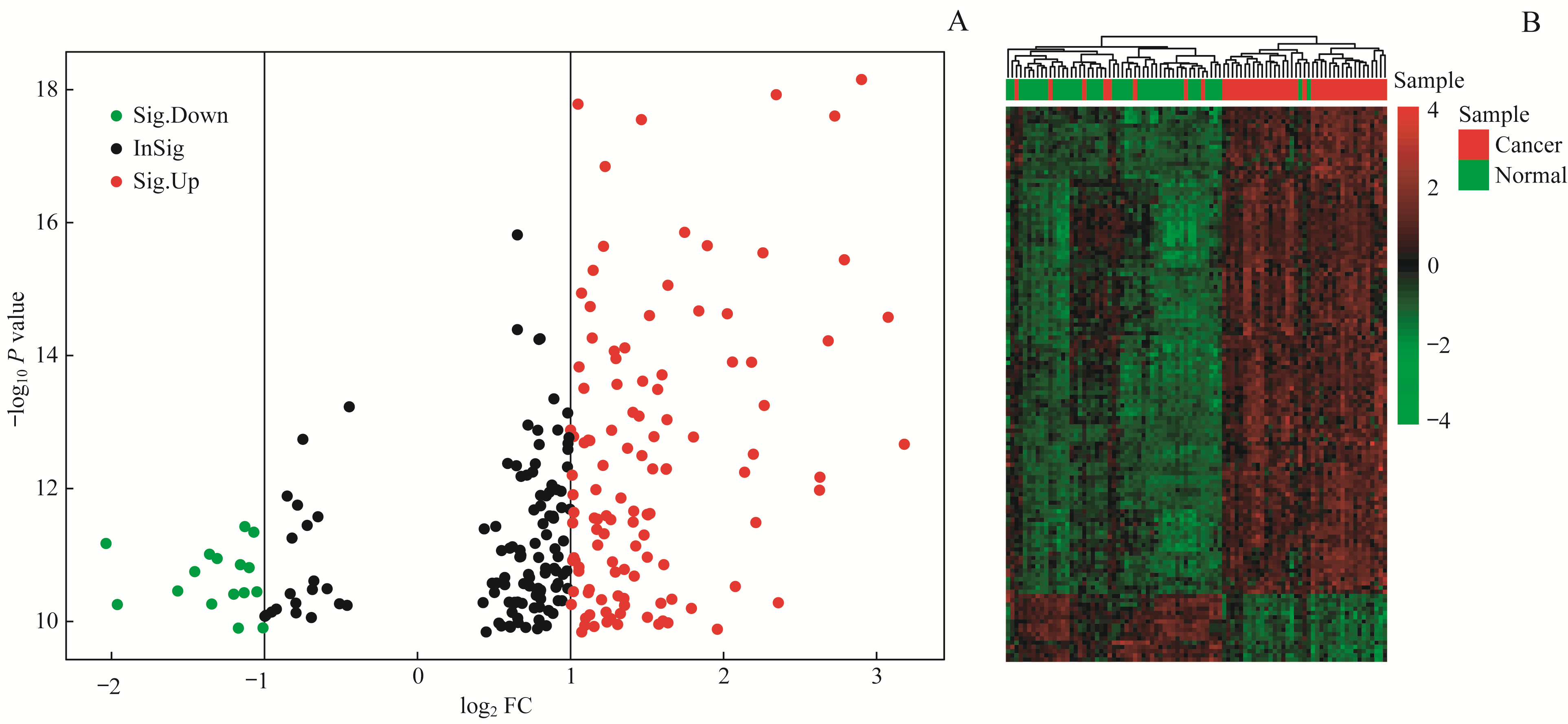
Fig 1 Visual analysis of the DEGs in pancreatic cancer and adjacent tissues
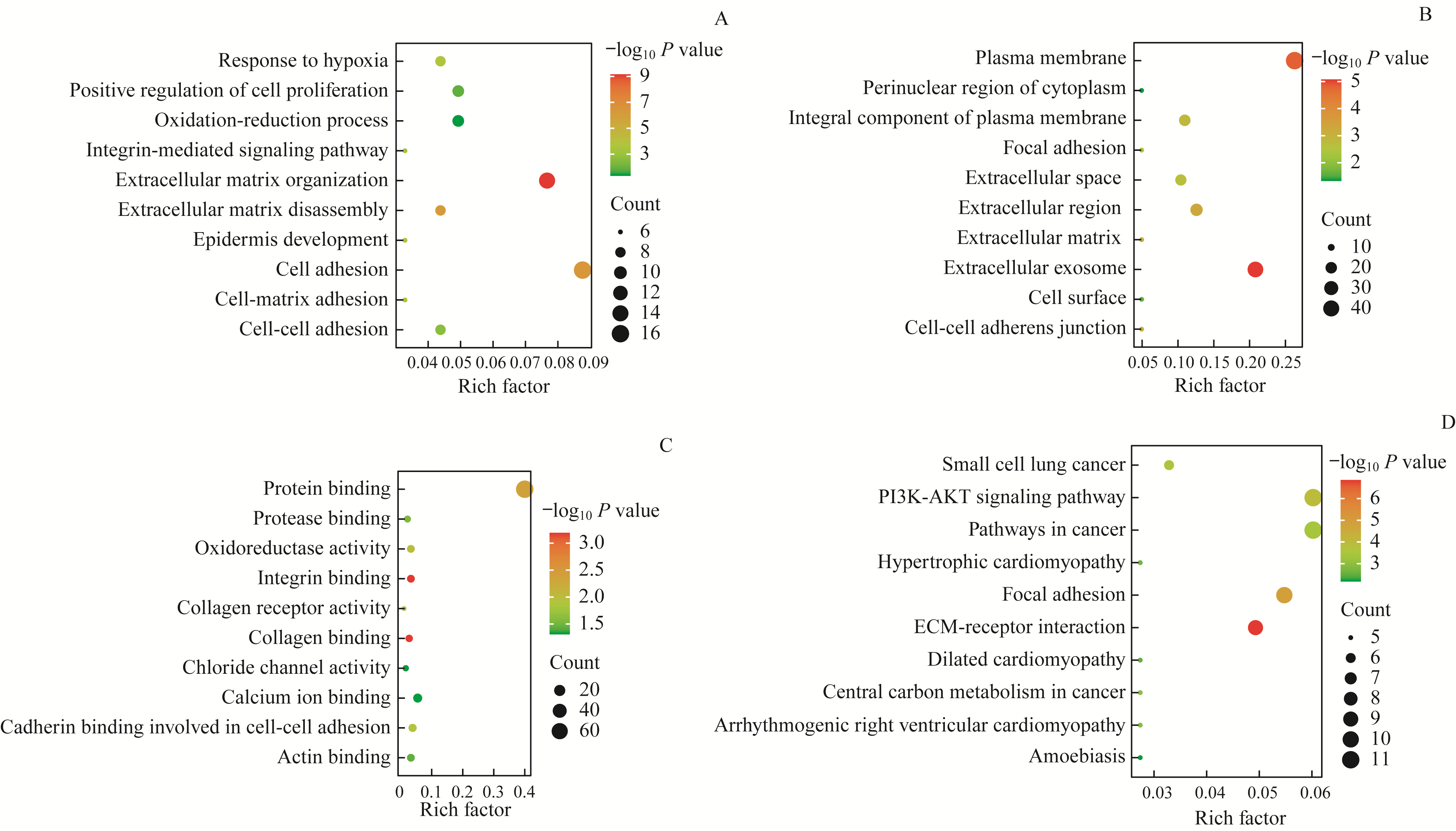
Fig 2 Enrichment analysis of GO function and KEGG pathway of DEGs

Fig 3 PPI network diagram of DEGs corresponding proteins
| Gene | Degree | Biological characteristic |
|---|---|---|
| FN1 | 24 | Involved in cell adhesion, cell movement, wound healing and maintenance of cell morphology |
| MET | 15 | Regulating cell proliferation, dispersion, proliferation and differentiation |
| LAMB3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| LAMA3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| ITGA3 | 11 | Involved in cell adhesion, invasion foot formation and matrix degradation |
Tab 1 Biological characteristics of the five DEGs in PPI network
| Gene | Degree | Biological characteristic |
|---|---|---|
| FN1 | 24 | Involved in cell adhesion, cell movement, wound healing and maintenance of cell morphology |
| MET | 15 | Regulating cell proliferation, dispersion, proliferation and differentiation |
| LAMB3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| LAMA3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| ITGA3 | 11 | Involved in cell adhesion, invasion foot formation and matrix degradation |
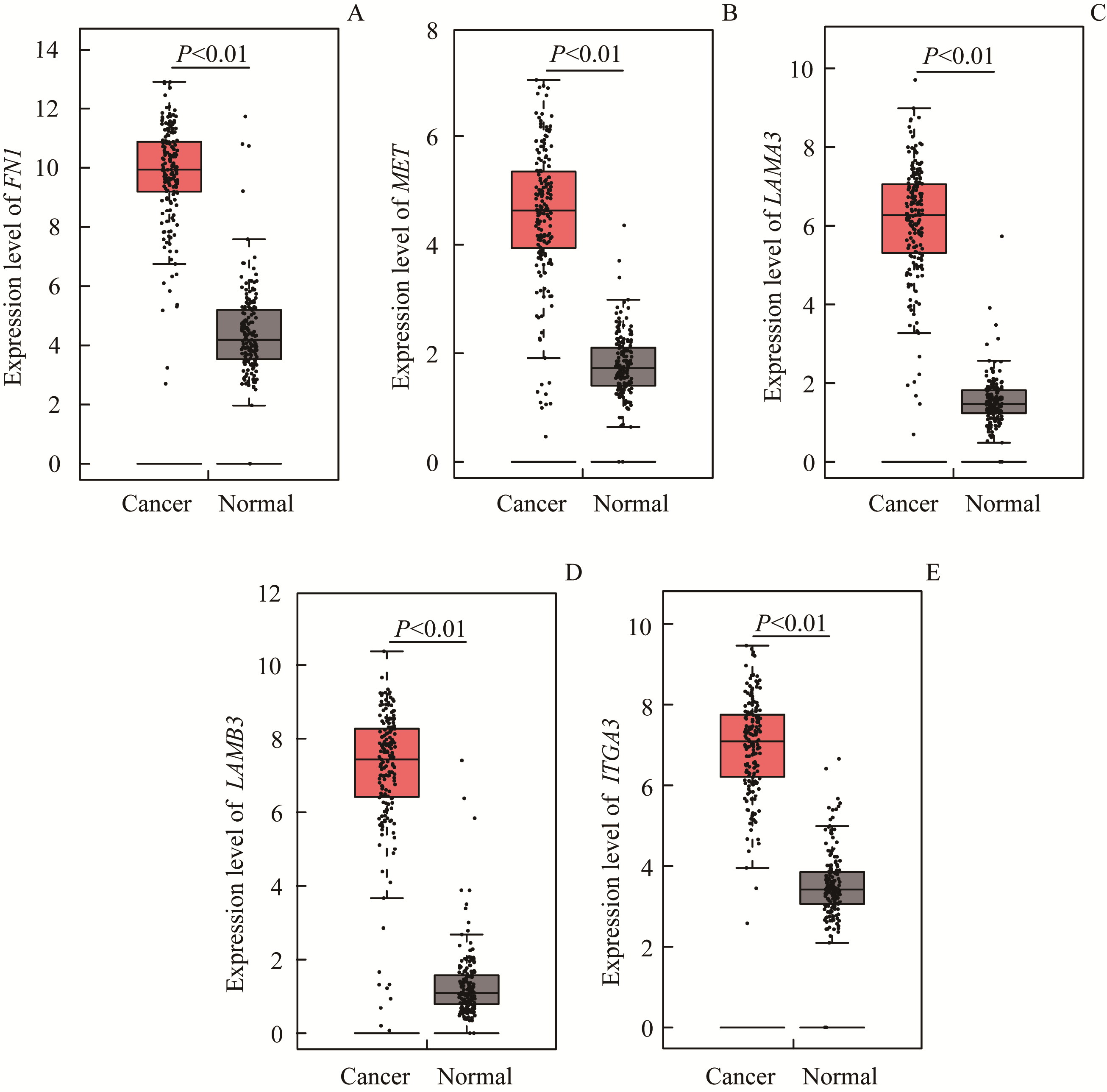
Fig 4 Expression of the five DEGs in 179 cases of pancreatic cancer tissues and 171 cases of adjacent normal tissues
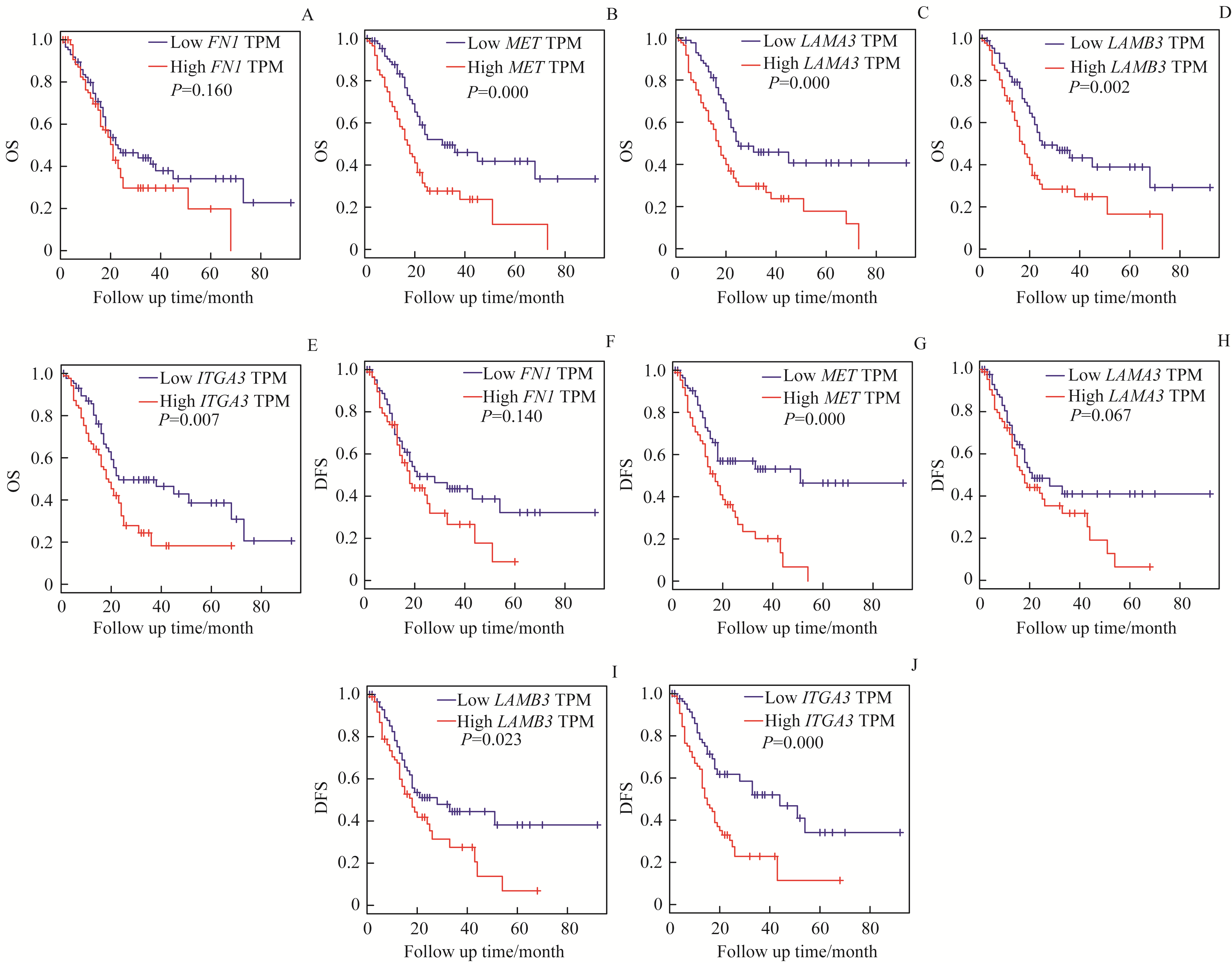
Fig 5 Prognostic significance of the five DEGs for pancreatic cancer patients assessed via Kaplan-Meier analysis
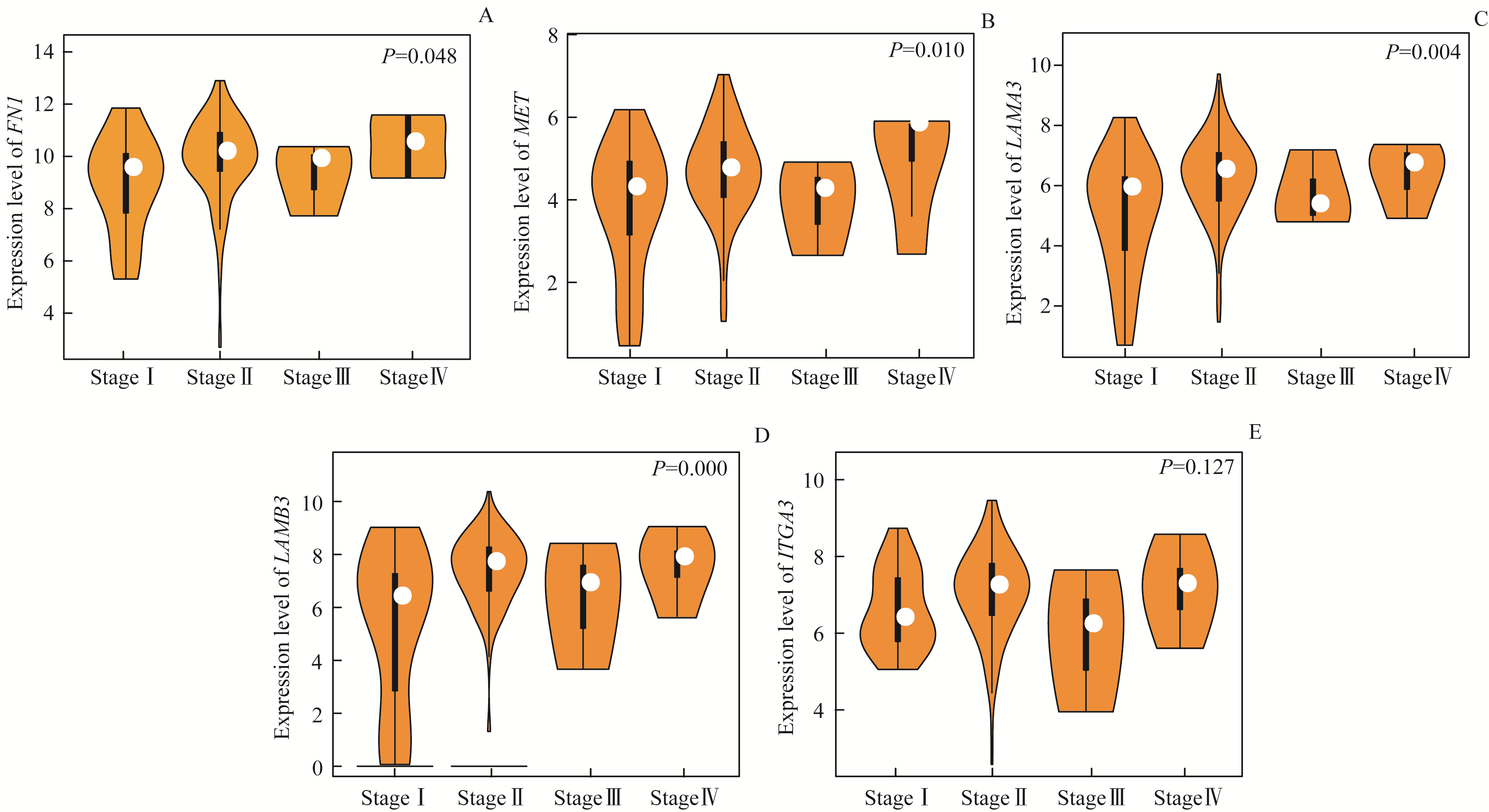
Fig 6 Expression of the five DEGs in pancreatic carcinoma with different pathological stages
| 1 | Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2018, 68(6): 394-424. |
| 2 | Ferlay J, Partensky C, Bray F. More deaths from pancreatic cancer than breast cancer in the EU by 2017[J]. Acta Oncol, 2016, 55(9/10): 1158-1160. |
| 3 | Lin QJ, Yang F, Jin C, et al. Current status and progress of pancreatic cancer in China[J]. World J Gastroenterol, 2015, 21(26): 7988-8003. |
| 4 | Mayo SC, Nathan H, Cameron JL, et al. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent[J]. Cancer, 2012, 118(10): 2674-2681. |
| 5 | de Sá MC, Simão ANC, de Medeiros FA, et al. Cell adhesion molecules and plasminogen activator inhibitor type-1 (PAI-1) in patients with rheumatoid arthritis: influence of metabolic syndrome[J]. Clin Exp Med, 2018, 18(4): 495-504. |
| 6 | Bendas G, Borsig L. Heparanase in cancer metastasis: heparin as a potential inhibitor of cell adhesion molecules[J]. Adv Exp Med Biol, 2020, 1221: 309-329. |
| 7 | Bergmann F, Wandschneider F, Sipos B, et al. Elevated L1CAM expression in precursor lesions and primary and metastastic tissues of pancreatic ductal adenocarcinoma[J]. Oncol Rep, 2010, 24(4): 909-915. |
| 8 | Geismann C, Morscheck M, Koch D, et al. Up-regulation of L1CAM in pancreatic duct cells is transforming growth factor β1- and slug-dependent: role in malignant transformation of pancreatic cancer[J]. Cancer Res, 2009, 69(10): 4517-4526. |
| 9 | Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment[J]. Annu Rev Med, 2016, 67: 11-28. |
| 10 | Liang H, Mokrani A, Chisomo-Kasiya H, et al. Dietary leucine affects glucose metabolism and lipogenesis involved in TOR/PI3K/Akt signaling pathway for juvenile blunt snout bream Megalobrama amblycephala[J]. Fish Physiol Biochem, 2019, 45(2): 719-732. |
| 11 | Polivka J, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway[J]. Pharmacol Ther, 2014, 142(2): 164-175. |
| 12 | Akinleye A, Avvaru P, Furqan M, et al. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics[J]. J Hematol Oncol, 2013, 6(1): 88. |
| 13 | Sierra JR, Tsao MS. C-MET as a potential therapeutic target and biomarker in cancer[J]. Ther Adv Med Oncol, 2011, 3(1): S21-S35. |
| 14 | Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification[J]. J Thorac Oncol, 2011, 6(5): 942-946. |
| 15 | Li Y, Chen CQ, He YL, et al. Abnormal expression of E-cadherin in tumor cells is associated with poor prognosis of gastric carcinoma[J]. J Surg Oncol, 2012, 106(3): 304-310. |
| 16 | di Renzo MF, Olivero M, Giacomini A, et al. Overexpression and amplification of themet/HGF receptor gene during the progression of colorectal cancer[J]. Clin Cancer Res, 1995, 1(2): 147-154. |
| 17 | Garcia S, Dalès JP, Jacquemier J, et al. C-Met overexpression in inflammatory breast carcinomas: automated quantification on tissue microarrays[J]. Br J Cancer, 2007, 96(2): 329-335. |
| 18 | Radaeva S, Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: a link between biliary intestinal Metaplasia and mucin-producing cholangiocarcinoma[J]. Hepatology, 1999, 29(5): 1453-1462. |
| 19 | di Renzo MF, Poulsom R, Olivero M, et al. Expression of the Met/hepatocyte growth factor receptor in human pancreatic cancer[J]. Cancer Res, 1995, 55(5): 1129-1138. |
| 20 | Neuzillet C, Couvelard A, Tijeras-Raballand A, et al. High c-Met expression in stage Ⅰ‒Ⅱ pancreatic adenocarcinoma: proposal for an immunostaining scoring method and correlation with poor prognosis[J]. Histopathology, 2015, 67(5): 664-676. |
| 21 | Hervieu A, Kermorgant S. The role of PI3K in Met driven cancer: a recap[J]. Front Mol Biosci, 2018, 5: 86. |
| 22 | Marinkovich MP. Tumour microenvironment: laminin 332 in squamous-cell carcinoma[J]. Nat Rev Cancer, 2007, 7(5): 370-380. |
| 23 | Stemmler S, Parwez Q, Petrasch-Parwez E, et al. Association of variation in the LAMA3 gene, encoding the α-chain of laminin 5, with atopic dermatitis in a German case-control cohort[J]. BMC Dermatol, 2014, 14: 17. |
| 24 | Castro BGR, Dos Reis R, Cintra GF, et al. Predictive factors for surgical morbidities and adjuvant chemotherapy delay for advanced ovarian cancer patients treated by primary debulking surgery or interval debulking surgery[J]. Int J Gynecol Cancer, 2018, 28(8): 1520-1528. |
| 25 | Lincoln V, Cogan J, Hou Y, et al. Gentamicin induces LAMB3 nonsense mutation readthrough and restores functional laminin 332 in junctional epidermolysis bullosa[J]. PNAS, 2018, 115(28): E6536-E6545. |
| 26 | Svoboda M, Hlobilová M, Marešová M, et al. Comparison of suction blistering and tape stripping for analysis of epidermal genes, proteins and lipids[J]. Arch Dermatol Res, 2017, 309(9): 757-765. |
| 27 | Wang YH, Jin YX, Bhandari A, et al. Upregulated LAMB3 increases proliferation and metastasis in thyroid cancer[J]. Onco Targets Ther, 2018, 11: 37-46. |
| 28 | Pan ZF, Li L, Fang QL, et al. Analysis of dynamic molecular networks for pancreatic ductal adenocarcinoma progression[J]. Cancer Cell Int, 2018, 18: 214. |
| 29 | Jung SN, Lim HS, Liu LH, et al. LAMB3 mediates metastatic tumor behavior in papillary thyroid cancer by regulating c-MET/Akt signals[J]. Sci Rep, 2018, 8(1): 2718. |
| 30 | Huang WJ, Gu JY, Tao T, et al. MiR-24-3p inhibits the progression of pancreatic ductal adenocarcinoma through LAMB3 downregulation[J]. Front Oncol, 2019, 9: 1499. |
| 31 | Zhang H, Pan YZ, Cheung M, et al. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway[J]. Cell Death Dis, 2019, 10(3): 230. |
| [1] | Rui-jie GENG, Lin YAO, Xin-xin HUANG, Shun-ying YU, Cheng-mei YUAN, Wu HONG, Qin-yu LÜ, Qing-zhong WANG, Zheng-hui YI, Yi-ru FANG. Identification of differentially expressed gene modules in major depressive disorder based on weighted gene co-expression network analysis [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(6): 724-731. |
| [2] | Yin LIU, Tao YANG, Yu-sai XIE, Yu-zhu WANG. Screening of key genes and pathways involved in lupus nephritis based on GEO database [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(6): 749-755. |
| [3] | Ling-ling LI, Qian LI, Ming-yu LI, Zheng LIU, Qian-cheng SHEN. Analysis of tumor immune-related differentially expressed genes in adults and children with acute myeloid leukemia [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(5): 579-587. |
| [4] | Qi-sheng GU, Mi-li ZHANG, Can CAO, Ji-kun LI. Association of alternative splicing and tumor immune in gastric cancer based on TCGA data set [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(4): 448-458. |
| [5] | Ren-yan WU, Xiao-lin GUO, Deng-li HONG, Lei CHEN. Identification of potential therapeutic target genes in pediatric acute leukemia of ambiguous lineage based on bioinformatics analysis [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(3): 320-327. |
| [6] | Yan-ru MA, Lin-hua JI, Tian-ying TONG, Yu-qing YAN, Chao-qin SHEN, Xin-yu ZHANG, Ying-ying CAO, Jie HONG, Hao-yan CHEN. Establishment and validation of prognostic prediction model of colorectal cancer based on single-cell RNA sequencing [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(2): 159-165. |
| [7] | Guo-qin HU, Qin-yu LÜ, Jing ZHAO, Ming-huan ZHU, Shun-ying YU, Zheng-hui YI, Jian CHEN. Association study of non-coding variant of NOS1AP gene with schizophrenia [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(1): 29-34. |
| [8] | Yan TONG, Jun-yan FANG, Hai DENG, A-hui SONG, Pu LI, Ying-li LIU. Different expression levels of exosomal miR-200a in peritoneal dialysis effluent from patients with different peritoneal transport characteristics and prediction of its biological function [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(1): 42-48. |
| [9] | GAO Jing-ze, WU Xia. CXCL9 mRNA in ovarian tumor tissue and its relations with prognosis and characteristics of immune microenvironment [J]. , 2020, 40(4): 457-. |
| [10] | DING Lei, GAO Cai-xia, LIU Zhao-yuan, CHEN Lei. Evaluation of a custom transcriptome sequencing library construction reagent with a small amount of cell input [J]. , 2020, 40(4): 472-. |
| [11] | CHEN Si, LIU Chun-liang, ZHAO Qian, SUN Hai-peng, LIU Yun-xia. Identification of hub genes and key pathways in breast cancersurvival-based bioinformatics analysis [J]. , 2020, 40(3): 294-. |
| [12] | XIA Shou-bing, XU Chun-jie, JIANG Chun-hui, GU Lei, SUN Long-ci, XU Qing. Bioinformatics analysis of ulcerative colitis and its malignant complications and screening of potential therapeutic drugs [J]. , 2020, 40(3): 317-. |
| [13] | LI Qian, GAO Jing-ze, LI Yun, SONG Kun, SHEN Qian-cheng. Bioinformatics analysis of esophageal squamous cell carcinoma genomic chip and prediction of targeted drug [J]. , 2020, 40(2): 194-. |
| [14] | ZHANG Wei-ran1, 2, LIN Xue-feng3, LI Xin2, ZHANG Hao2, WANG Meng2, SUN Wei2, HAN Xing-peng2, SUN Da-qiang1, 4. Transcriptional identification of potential biomarkers of lung adenocarcinoma [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2020, 40(12): 1598-1606. |
| [15] | YU Meng-na1, YANG Biao2, YANG Li-zhen1. Prediction and bioinformatics analysis of hsa-miR-223-3p target genes related to diabetes mellitus [J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2020, 40(11): 1477-1484. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||