Journal of Shanghai Jiao Tong University (Medical Science) ›› 2026, Vol. 46 ›› Issue (3): 291-300.doi: 10.3969/j.issn.1674-8115.2026.03.003
• Basic research • Previous Articles
Chen Jiayu, Zhang Huili( )
)
Received:2025-11-04
Accepted:2025-12-10
Online:2026-03-28
Published:2026-03-30
Contact:
Zhang Huili
E-mail:huilizhang815@163.com
Supported by:CLC Number:
Chen Jiayu, Zhang Huili. Role and mechanism of fibroblast mitochondrial dysfunction in pulmonary arterial hypertension[J]. Journal of Shanghai Jiao Tong University (Medical Science), 2026, 46(3): 291-300.
Add to citation manager EndNote|Ris|BibTeX
URL: https://xuebao.shsmu.edu.cn/EN/10.3969/j.issn.1674-8115.2026.03.003
| Primer name | Sequences (5′→3′) |
|---|---|
| α-tubulin | |
| Forward | CTGATGTATGCCAAGCGTGC |
| Reverse | ATCCTTCTCTAGGGCAGCCA |
| Col1a1 | |
| Forward | CCAGCCGCAAAGAGTCTACAT |
| Reverse | ATGTCTTCTTGGCCATGCGT |
| Col3a1 | |
| Forward | GCCTACATGGATCAGGCCAA |
| Reverse | CATGGCCTTGCGTGTTTGAT |
Tab 1 Primer sequences for PCR
| Primer name | Sequences (5′→3′) |
|---|---|
| α-tubulin | |
| Forward | CTGATGTATGCCAAGCGTGC |
| Reverse | ATCCTTCTCTAGGGCAGCCA |
| Col1a1 | |
| Forward | CCAGCCGCAAAGAGTCTACAT |
| Reverse | ATGTCTTCTTGGCCATGCGT |
| Col3a1 | |
| Forward | GCCTACATGGATCAGGCCAA |
| Reverse | CATGGCCTTGCGTGTTTGAT |
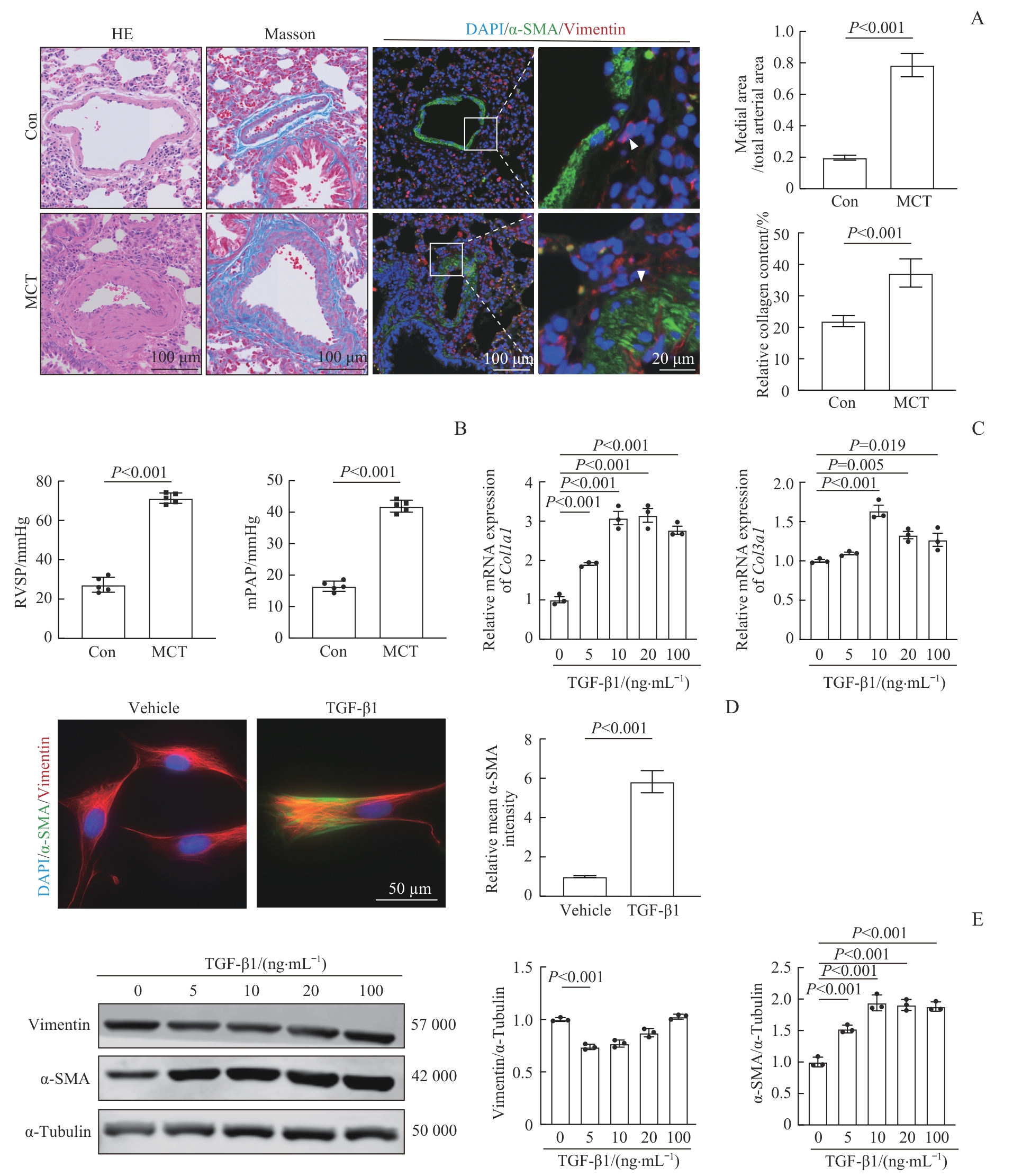
Fig 1 Establishment of the PAH model and FMT in fibroblasts
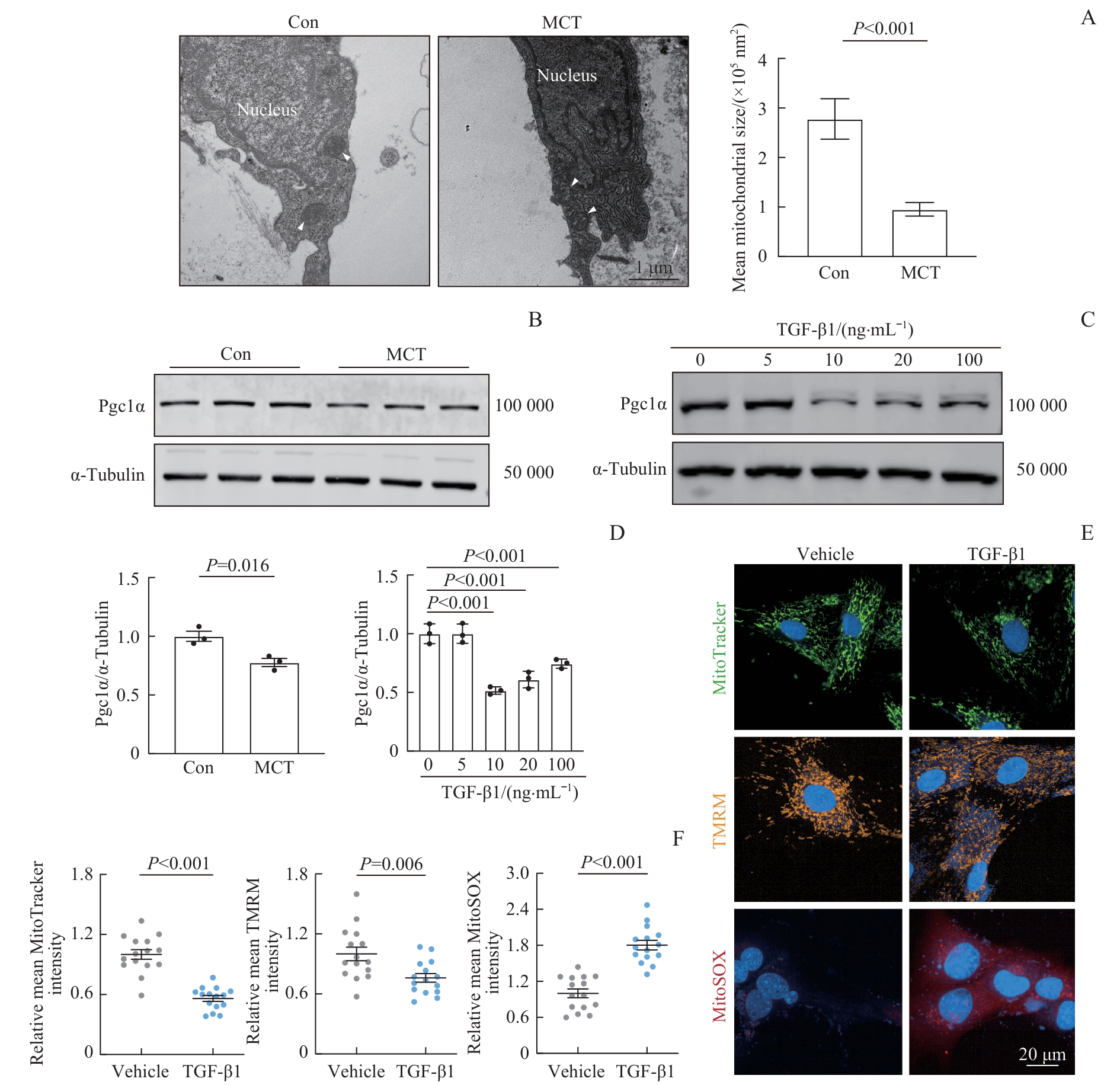
Fig 2 Mitochondrial dysfunction in fibroblasts in the PAH model
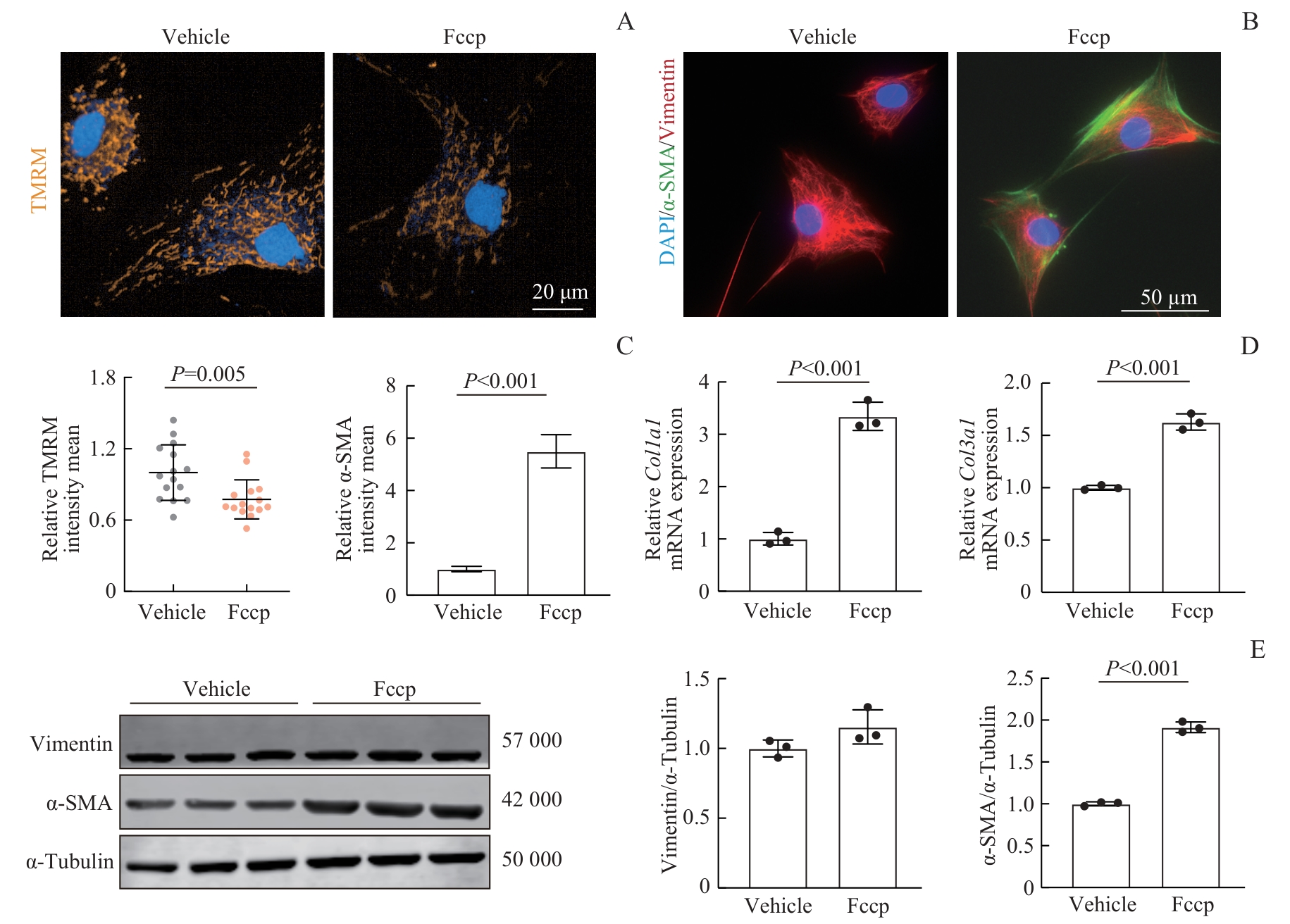
Fig 3 Fccp-induced mitochondrial dysfunction and FMT
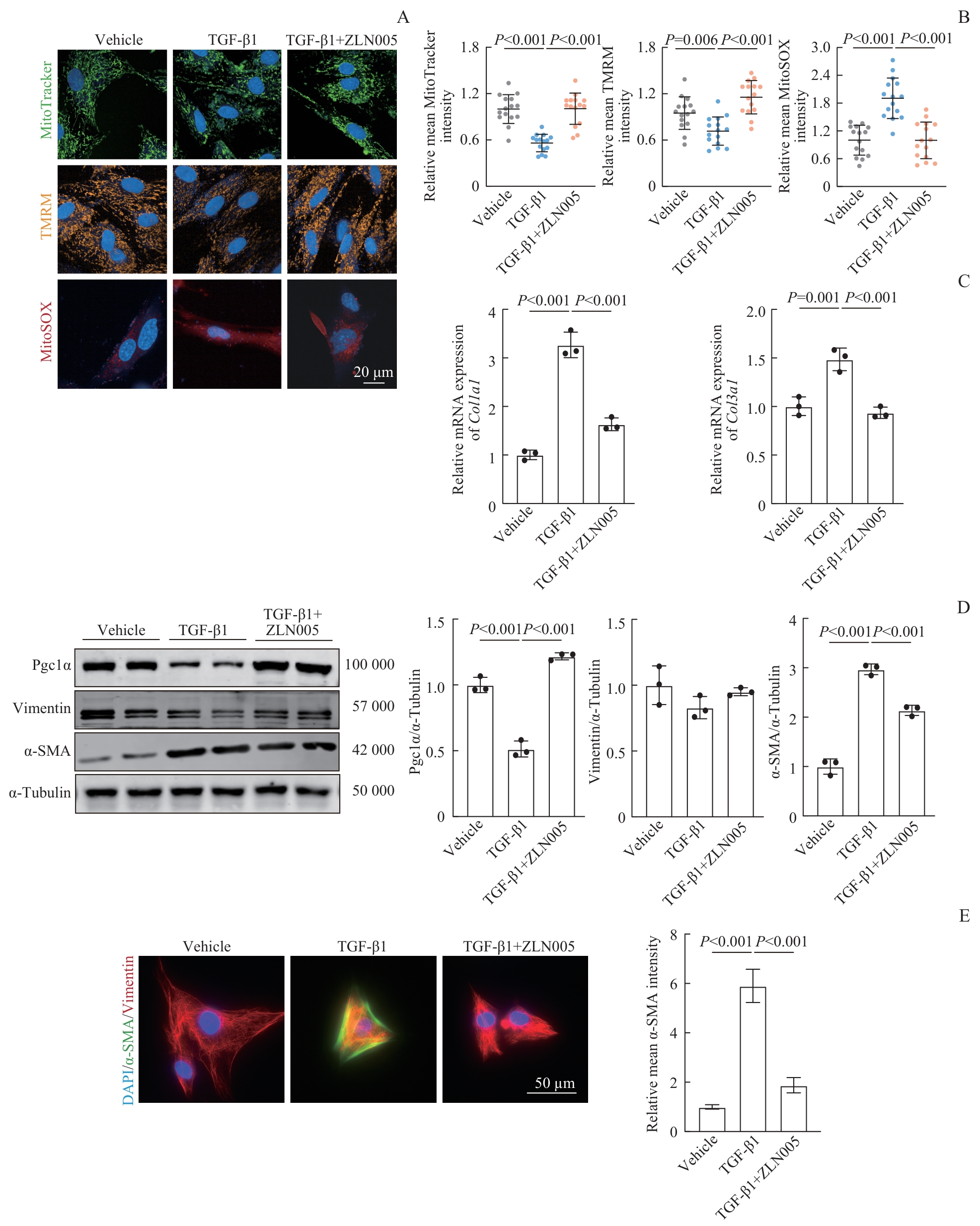
Fig 4 ZLN005-mediated improvement in mitochondrial function and suppression of FMT
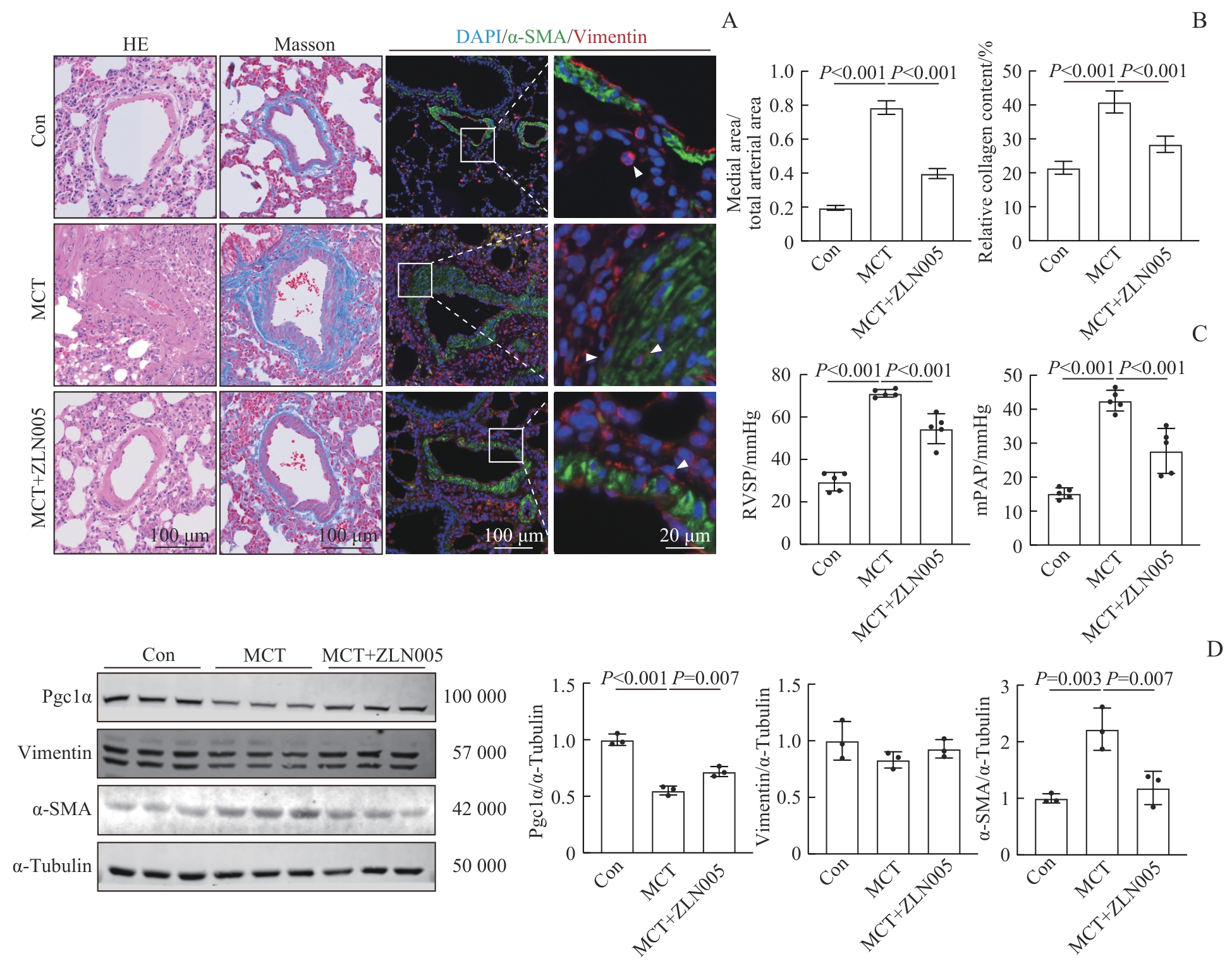
Fig 5 Amelioration of PAH in rats induced by ZLN005
| [1] | Mocumbi A, Humbert M, Saxena A, et al. Pulmonary hypertension[J]. Nat Rev Dis Primers, 2024, 10: 1. |
| [2] | Strange G, Playford D, Stewart S, et al. Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort[J]. Heart, 2012, 98(24): 1805-1811. |
| [3] | Hassoun P M. Pulmonary arterial hypertension[J]. N Engl J Med, 2021, 385(25): 2361-2376. |
| [4] | Guignabert C, Aman J, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: current insights and future directions[J]. Eur Respir J, 2024, 64(4): 2401095. |
| [5] | Thenappan T, Ormiston M L, Ryan J J, et al. Pulmonary arterial hypertension: pathogenesis and clinical management[J]. BMJ, 2018, 360: j5492. |
| [6] | Liang B H, Lin W C, Tang Y Y, et al. Selenium supplementation elevated SELENBP1 to inhibit fibroblast activation in pulmonary arterial hypertension[J]. iScience, 2024, 27(10): 111036. |
| [7] | Rachedi N S, Tang Y, Tai Y Y, et al. Dietary intake and glutamine-serine metabolism control pathologic vascular stiffness[J]. Cell Metab, 2024, 36(6): 1335-1350.e8. |
| [8] | Zhang H, Li M, Hu C J, et al. Fibroblasts in pulmonary hypertension: roles and molecular mechanisms[J]. Cells, 2024, 13(11): 914. |
| [9] | Kurose H. Cardiac fibrosis and fibroblasts[J]. Cells, 2021, 10(7): 1716. |
| [10] | Gibb A A, Lazaropoulos M P, Elrod J W. Myofibroblasts and fibrosis: mitochondrial and metabolic control of cellular differentiation[J]. Circ Res, 2020, 127(3): 427-447. |
| [11] | Seok B G, Lee S, Jin S H, et al. Inhibiting mitoNEET restores mitochondrial redox homeostasis and attenuates myofibroblast differentiation[J]. Free Radic Biol Med, 2025, 238: 293-302. |
| [12] | Halling J F, Pilegaard H. PGC-1α-mediated regulation of mitochondrial function and physiological implications[J]. Appl Physiol Nutr Metab, 2020, 45(9): 927-936. |
| [13] | Abu Shelbayeh O, Arroum T, Morris S, et al. PGC-1α is a master regulator of mitochondrial lifecycle and ROS stress response[J]. Antioxidants (Basel), 2023, 12(5): 1075. |
| [14] | Evans C E, Cober N D, Dai Z Y, et al. Endothelial cells in the pathogenesis of pulmonary arterial hypertension[J]. Eur Respir J, 2021, 58(3): 2003957. |
| [15] | Shen H, Gao Y, Ge D D, et al. BRCC3 regulation of ALK2 in vascular smooth muscle cells: implication in pulmonary hypertension[J]. Circulation, 2024, 150(2): 132-150. |
| [16] | MacKay C D A, Jadli A S, Fedak P W M, et al. Adventitial fibroblasts in aortic aneurysm: unraveling pathogenic contributions to vascular disease[J]. Diagnostics (Basel), 2022, 12(4): 871. |
| [17] | Younesi F S, Miller A E, Barker T H, et al. Fibroblast and myofibroblast activation in normal tissue repair and fibrosis[J]. Nat Rev Mol Cell Biol, 2024, 25(8): 617-638. |
| [18] | Wang A, Valdez-Jasso D. Cellular mechanosignaling in pulmonary arterial hypertension[J]. Biophys Rev, 2021, 13(5): 747-756. |
| [19] | Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives[J]. Eur Respir J, 2019, 53(1): 1801887. |
| [20] | Wang D R, Zhang H, Li M, et al. microRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts[J]. Circ Res, 2014, 114(1): 67-78. |
| [21] | Chelladurai P, Boucherat O, Stenmark K, et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy[J]. Br J Pharmacol, 2021, 178(1): 54-71. |
| [22] | Khatun J, Gelles J D, Chipuk J E. Dynamic death decisions: how mitochondrial dynamics shape cellular commitment to apoptosis and ferroptosis[J]. Dev Cell, 2024, 59(19): 2549-2565. |
| [23] | Newmeyer D D, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death[J]. Cell, 2003, 112(4): 481-490. |
| [24] | Su C T, See D H W, Huang Y J, et al. LTBP4 protects against renal fibrosis via mitochondrial and vascular impacts[J]. Circ Res, 2023, 133(1): 71-85. |
| [25] | Peng F, Liao M R, Jin W K, et al. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis[J]. Signal Transduct Target Ther, 2024, 9(1): 133. |
| [26] | Ahangari F, Price N L, Malik S, et al. microRNA-33 deficiency in macrophages enhances autophagy, improves mitochondrial homeostasis, and protects against lung fibrosis[J]. JCI Insight, 2023, 8(4): e158100. |
| [27] | Zhang T, Liu C F, Zhang T N, et al. Overexpression of peroxisome proliferator-activated receptor γ coactivator 1-α protects cardiomyocytes from lipopolysaccharide-induced mitochondrial damage and apoptosis[J]. Inflammation, 2020, 43(5): 1806-1820. |
| [28] | Humeres C, Shinde A V, Tuleta I, et al. Fibroblast Smad7 induction protects the remodeling pressure-overloaded heart[J]. Circ Res, 2024, 135(3): 453-469. |
| [29] | Tie Y, Tang F, Peng D D, et al. TGF-beta signal transduction: biology, function and therapy for diseases[J]. Mol Biomed, 2022, 3(1): 45. |
| [30] | Kumar A P, Puthussery D T. Regulation of PPAR-γ coactivator-1α and its implication in mitochondrial function and neurodegenerative diseases[J]. Ageing Res Rev, 2025, 112: 102887. |
| [31] | Huang T, Zhang T Y, Gao J Q. Targeted mitochondrial delivery: a therapeutic new era for disease treatment[J]. J Control Release, 2022, 343: 89-106. |
| [32] | Tohme C, Haykal T, Yang R Q, et al. ZLN005, a PGC-1α activator, protects the liver against ischemia-reperfusion injury and the progression of hepatic metastases[J]. Cells, 2024, 13(17): 1448. |
| [33] | Zhu P F, Ma H J, Cui S C, et al. ZLN005 alleviates in vivo and in vitro renal fibrosis via PGC-1α-mediated mitochondrial homeostasis[J]. Pharmaceuticals (Basel), 2022, 15(4): 434. |
| [34] | Hsu C H, Roan J N, Fang S Y, et al. Transplantation of viable mitochondria improves right ventricular performance and pulmonary artery remodeling in rats with pulmonary arterial hypertension[J]. J Thorac Cardiovasc Surg, 2022, 163(5): e361-e373. |
| [35] | Gollihue J L, Patel S P, Eldahan K C, et al. Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury[J]. J Neurotrauma, 2018, 35(15): 1800-1818. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||