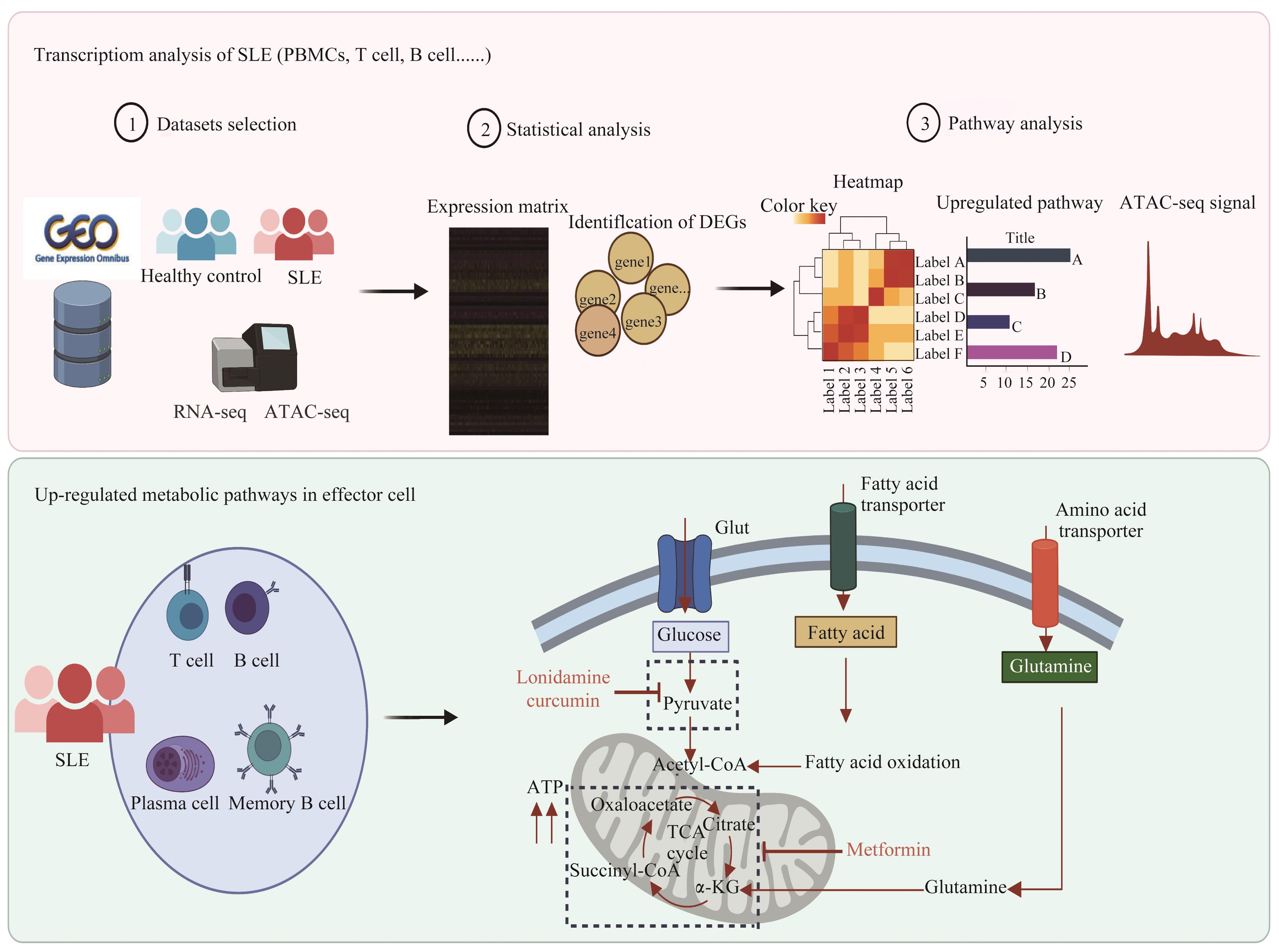
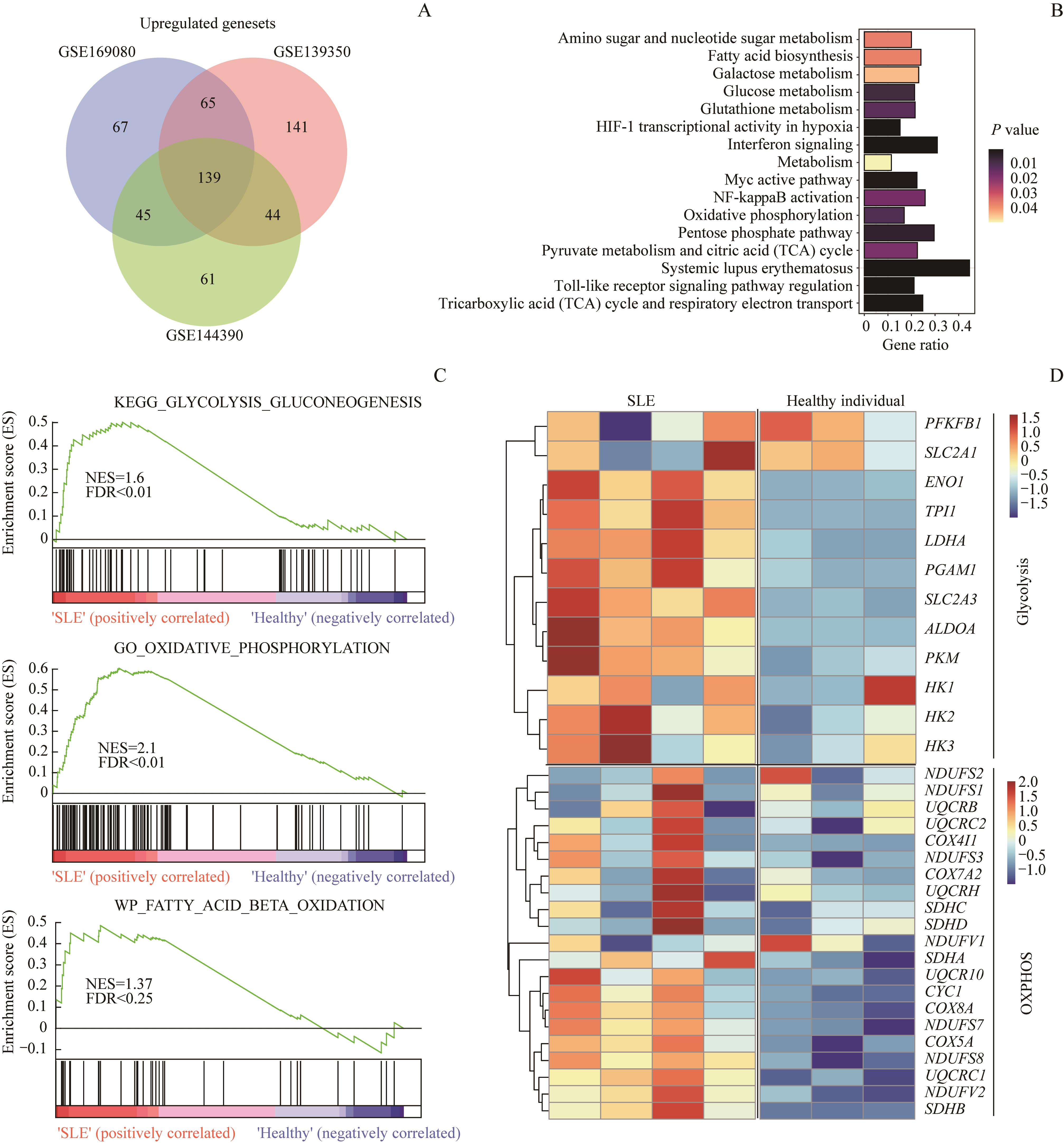
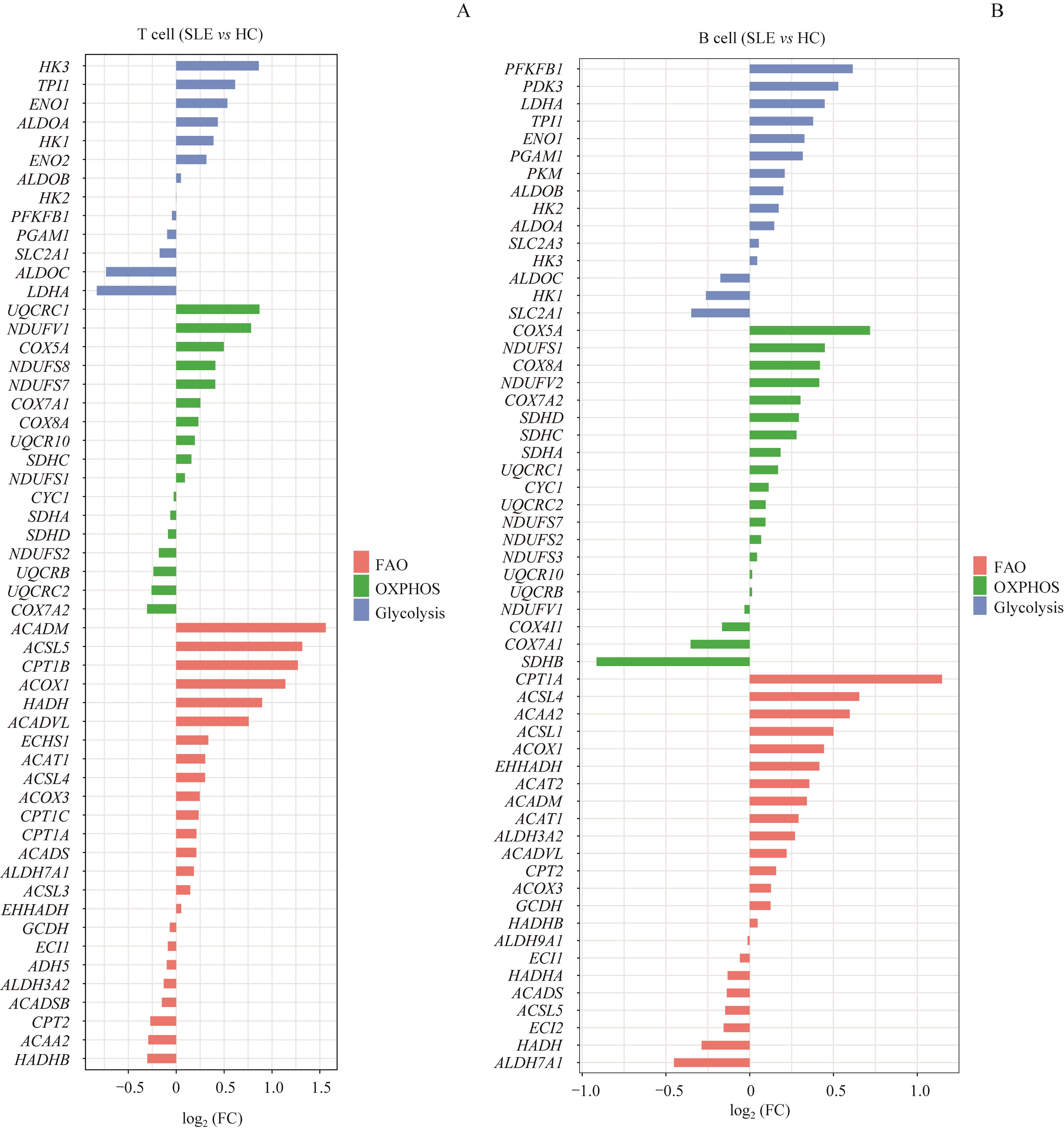
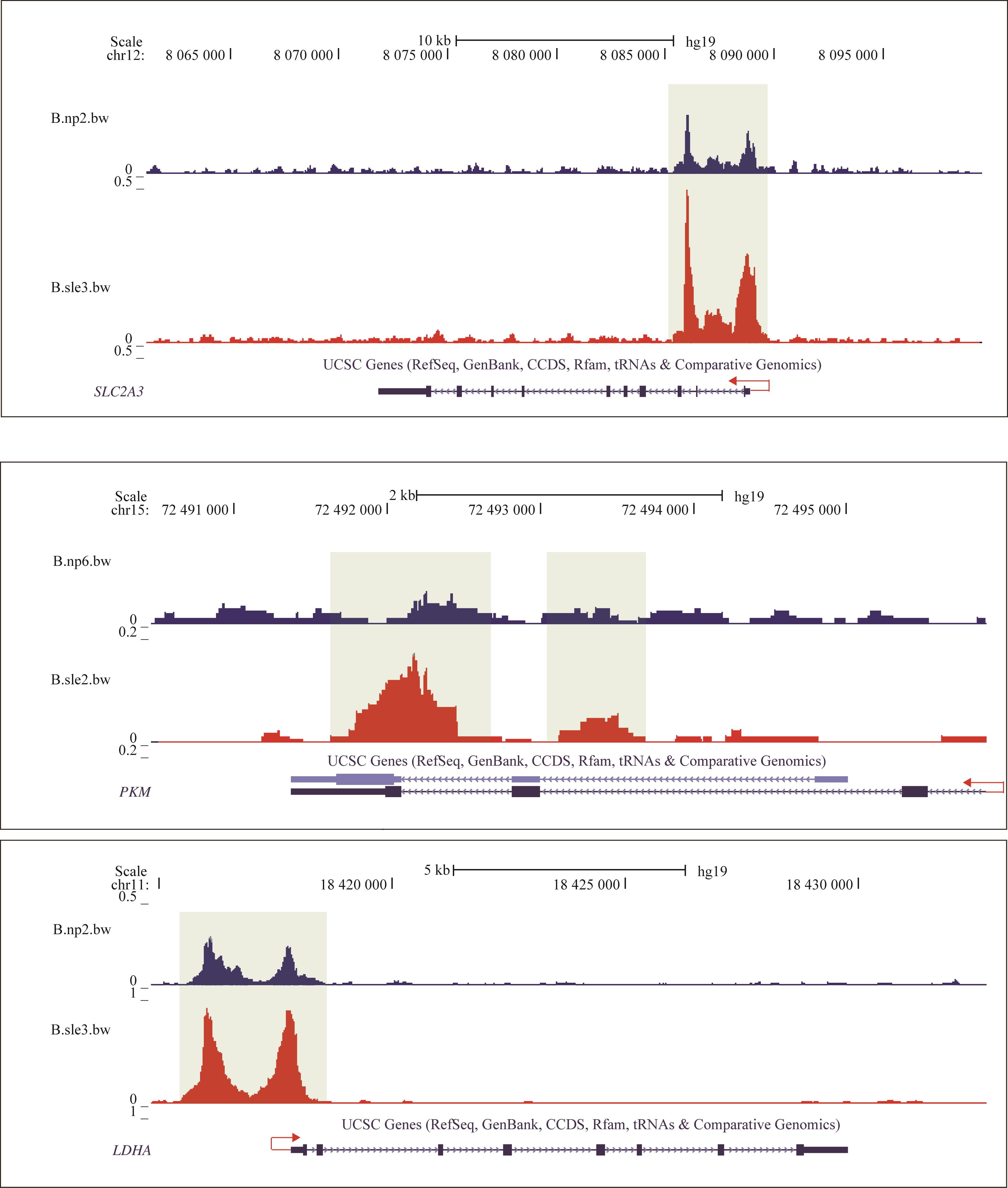
| 1 |
DÖRNER T, FURIE R. Novel paradigms in systemic lupus erythematosus[J]. Lancet, 2019, 393(10188): 2344-2358.
|
| 2 |
HE J Q, CHAN T L, HONG X P, et al. Microbiome and metabolome analyses reveal the disruption of lipid metabolism in systemic lupus erythematosus[J]. Front Immunol, 2020, 11: 1703.
|
| 3 |
ZHANG L S, QING P Y, YANG H, et al. Gut microbiome and metabolites in systemic lupus erythematosus: link, mechanisms and intervention[J]. Front Immunol, 2021, 12: 686501.
|
| 4 |
CHOI S C, BROWN J, GONG M H, et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice[J]. Sci Transl Med, 2020, 12(551): eaax2220.
|
| 5 |
YIN Y M, CHOI S C, XU Z W, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus[J]. J Immunol, 2016, 196(1): 80-90.
|
| 6 |
YIN Y M, CHOI S C, XU Z W, et al. Normalization of CD4+ T cell metabolism reverses lupus[J]. Sci Transl Med, 2015, 7(274): 274ra18.
|
| 7 |
O'NEILL L A J, KISHTON R J, RATHMELL J. A guide to immunometabolism for immunologists[J]. Nat Rev Immunol, 2016, 16(9): 553-565.
|
| 8 |
WU T F, QIN X M, KUREPA Z, et al. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus[J]. J Clin Invest, 2007, 117(8): 2186-2196.
|
| 9 |
SARAVIA J, RAYNOR J L, CHAPMAN N M, et al. Signaling networks in immunometabolism[J]. Cell Res, 2020, 30(4): 328-342.
|
| 10 |
MAKOWSKI L, CHAIB M, RATHMELL J C. Immunometabolism: From basic mechanisms to translation[J]. Immunol Rev, 2020, 295(1): 5-14.
|
| 11 |
HOU G J, HARLEY I T W, LU X M, et al. SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression[J]. Nat Commun, 2021, 12(1): 135.
|
| 12 |
CHEN E Y, TAN C M, KOU Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool[J]. BMC Bioinformatics, 2013, 14: 128.
|
| 13 |
STURM G, FINOTELLO F, PETITPREZ F, et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology[J]. Bioinformatics, 2019, 35(14): i436-i445.
|
| 14 |
SHLOMCHIK M J, CRAFT J E, MAMULA M J. From T to B and back again: positive feedback in systemic autoimmune disease[J]. Nat Rev Immunol, 2001, 1(2): 147-153.
|
| 15 |
CORCORAN L M, NUTT S L. Long-lived plasma cells have a sweet tooth[J]. Immunity, 2016, 45(1): 3-5.
|
| 16 |
ZHOU X, MOTTA F, SELMI C, et al. Antibody glycosylation in autoimmune diseases[J]. Autoimmun Rev, 2021, 20(5): 102804.
|
| 17 |
REILY C, STEWART T J, RENFROW M B, et al. Glycosylation in health and disease[J]. Nat Rev Nephrol, 2019, 15(6): 346-366.
|
| 18 |
CORRADO M, PEARCE E L. Targeting memory T cell metabolism to improve immunity[J]. J Clin Invest, 2022, 132(1): e148546.
|
| 19 |
SUN L, ZHANG H, GAO P. Metabolic reprogramming and epigenetic modifications on the path to cancer[J]. Protein Cell, 2021: 2021May29.
|
| 20 |
ZHENG Y, WU C, YANG J M, et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer[J]. Signal Transduct Target Ther, 2020, 5(1): 53.
|
| 21 |
QIU R, YU X, WANG L, et al. Inhibition of glycolysis in pathogenic TH17 cells through targeting a miR-21-Peli1-c-rel pathway prevents autoimmunity[J]. J Immunol, 2020, 204(12): 3160-3170.
|
| 22 |
HUANG N, PERL A. Metabolism as a target for modulation in autoimmune diseases[J]. Trends Immunol, 2018, 39(7): 562-576.
|
| 23 |
ERIKSSON P, WALLIN P, SJÖWALL C. Clinical experience of sirolimus regarding efficacy and safety in systemic lupus erythematosus[J]. Front Pharmacol, 2019, 10: 82.
|
| 24 |
SUN F F, GENG S K, WANG H T, et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: post hoc analyses from two randomised trials[J]. Lupus Sci Med, 2020, 7(1): e000429.
|
 ), JIANG Yang, GU Shuangshuang, DAI Dai, SHEN Nan(
), JIANG Yang, GU Shuangshuang, DAI Dai, SHEN Nan(