

乳腺癌是女性最常见的癌症之一,也是女性癌症相关死亡的第二大原因[1]。据统计,2022年全球女性乳腺癌新发病例数约占所有新诊断癌症数的31%[1],位居女性恶性肿瘤第1位,因其死亡的人数估计占女性癌症相关死亡总人数的15%。近年来,临床上常采用免疫检查点抑制剂(immune checkpoint inhibitor,ICI)对乳腺癌患者进行治疗,该方法虽能大幅提升转移性乳腺癌患者的总生存率[2-4],但多数患者存在对免疫疗法无响应或在响应治疗后出现复发等问题,缓解率各不相同。因此,探究乳腺癌转移的分子机制并发现有潜力的预后生物标志物和分子靶点对于乳腺癌早期转移过程中的干预和治疗具有重要的科学意义。
研究[5-6]显示,乳腺癌的侵袭性和耐药性可能是由于其肿瘤微环境(tumor microenvironment,TME)的高度异质性所致。有研究发现,TME由肿瘤细胞、肿瘤浸润免疫细胞(tumor-infiltrating immune cell,TIIC)、细胞因子、趋化因子、细胞外基质等组成[7],且各组分间的相互作用是TME异质性的重要原因之一,可直接或间接地促进肿瘤转移[8-9]。其中,有研究[10]已证实TIIC的数量和活动状态可决定结直肠癌患者的生存时间。相关研究[11-13]显示,运输和招募免疫细胞到肿瘤组织中是启动和传递抗肿瘤免疫反应的重要环节,而运输和招募过程需通过特定的免疫细胞受体与趋化因子结合来介导。同时,肿瘤细胞也可以通过免疫检查点抑制T细胞的激活,从而逃避免疫杀伤。与免疫细胞低浸润肿瘤(“冷”肿瘤)的患者相比,免疫细胞高浸润肿瘤(“热”肿瘤)的患者预后较好[14],其更有可能从ICI中获益[15-16]。多种肿瘤类型的研究发现,CD8+ T细胞浸润比例的增加与患者更好的预后相关[14],且肿瘤抗原呈递和突变负荷是CD8+ T细胞浸润相关的重要因素之一[17-18]。在TME中,长期的抗原暴露刺激可导致CD8+ T细胞出现耗竭状态;在理想情况下,ICI则可逆转这种耗竭[19-20]。
在本研究中,我们利用癌症基因组图谱(The Cancer Genome Atlas,TCGA)、STRING、ImmPort和GEPIA2等多个数据库进行生物信息学分析,描述乳腺癌TME中免疫细胞的浸润特征,并筛选得到目的基因即C-X-C模体趋化因子配体9(C-X-C motif chemokine ligand 9,CXCL9)。而后,通过临床乳腺癌石蜡组织标本进行实验验证,探究CXCL9与乳腺癌TME中募集淋巴细胞并促进其发挥抗肿瘤免疫应答功能的相关性,旨在为CXCL9抑制乳腺癌进展的机制研究提供新的方向,并为其作为乳腺癌的良好预后标志物和治疗靶点提供新的证据。
1 资料与方法
1.1 公共数据来源
从TCGA数据库(
1.2 生物信息学数据分析
1.2.1 乳腺癌微环境中TIIC的相关分析
根据从TCGA数据库获取的转录组数据和患者临床信息,采用CIBERSORT反卷积算法评估乳腺癌微环境中22种TIIC亚群的比例,并通过R语言ggplot2包和pheatmap包绘制可视化聚类热图[21];采用Kruskal-Wallis检验分析TIIC亚群在4种分子分型间的占比差异。运用Kaplan-Meier(KM)生存曲线分析TIIC浸润水平与乳腺癌患者总生存期(overall survival,OS)的相关性。
1.2.2 目的基因CXCL9的筛选
根据TCGA数据库的转录组数据,使用R语言limma包分析乳腺癌和癌旁组织的差异表达基因(differentially expressed gene,DEG);将校正P<0.05、差异倍数(fold change,FC)的对数的绝对值(|log2(FC)|)>2作为DEG的筛选条件,取log2(FC)>2的上调基因作为基因集一。从ImmPort数据库(
1.2.3 CXCL9 mRNA的表达分析及其对患者的预后影响
根据从TCGA数据库获得的转录组数据和患者临床信息,在GEPIA2数据平台分析CXCL9 mRNA在不同类型的癌症及其对应的正常组织中的表达,利用Kruskal-Wallis检验分析CXCL9 mRNA在乳腺癌和癌旁组织、不同分子分型乳腺癌间的表达差异。根据CXCL9 mRNA表达量的中位数将患者分为高表达组和低表达组,并利用KM生存曲线分析其表达与乳腺癌患者OS的相关性。
1.2.4 蛋白质相互作用网络的构建
利用STRING (
1.2.5 CXCL9及其相关基因的GO功能分析和 KEGG通路分析
根据TCGA数据库获取的转录组数据,运用Spearman相关系数评估CXCL9和其他基因之间的相关性,并筛选出正相关系数中前150位的基因作为数据集二。使用R语言clusterProfiler包对数据集一和数据集二合并行基因本体数据库(Gene Ontology,GO)功能分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genome,KEGG)通路分析;其中,GO功能分析包括生物学过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)分析。设定P<0.05且富集因子>1.5为筛选标准。
1.2.6 CXCL9 mRNA表达与TIIC亚群的相关性分析及对患者OS影响
运用Spearman相关系数分析 CXCL9 mRNA表达与乳腺癌微环境中TIIC亚群、免疫检查点基因的相关性。根据CXCL9 mRNA表达量中位数和CD8+ T细胞浸润比例中位数分为4组,即组别1为CXCL9 mRNA低表达+CD8+ T细胞低浸润、组别2为CXCL9 mRNA低表达+CD8+ T细胞高浸润、组别3为CXCL9 mRNA高表达+CD8+ T细胞低浸润、组别4为CXCL9 mRNA高表达+CD8+ T细胞高浸润,利用KM生存曲线分析CXCL9 mRNA表达和CD8+ T细胞(TIIC亚群之一)浸润比例双因素对乳腺癌患者OS的影响。
1.3 组织标本收集
选择2016—2019年上海交通大学医学院附属第一人民医院的60例乳腺癌患者的石蜡组织样本(其中癌组织60例、癌旁组织40例),将其制作成组织芯片;采集患者的相关临床信息,包括肿瘤大小、分子分型、病理分期、生存时间等。
1.4 主要试剂及仪器
二甲苯和乙醇(国药集团化学试剂有限公司),通用型强力抗原修复液(上海碧云天生物技术有限公司),多重荧光免疫组织化学试剂盒(OpalTM 7-Color Manual IHC Kit,Akoya Biosciences,美国),CD8α抗体(Proteintech,美国),CXCL9抗体(Affinity,美国),CK7抗体、CD68抗体、CD11c抗体(Abcam,英国)。光学显微镜(Nikon,日本),全自动定量病理成像分析系统(PerkinElmer Vectra PolarisTM,美国)。
1.5 实验方法
1.5.1 CXCL9表达水平及其与CD8+ T细胞浸润的相关性分析
采用免疫组织化学染色(immunohistochemistry staining,IHC)对组织芯片中的乳腺癌和癌旁组织进行分析,即将芯片经65 ℃烘烤60 min、二甲苯脱蜡、乙醇梯度浓度水化后,用通用型强力抗原修复液水浴修复20 min,自然冷却至室温。采用内源性生物素封闭液进行封闭后,一抗(CD8α抗体、CXCL9抗体的工作浓度分别为1∶20 000、1∶200)4 ℃孵育过夜。次日使用二抗增强液、增强酶标山羊抗小鼠/兔IgG聚合物先后分别孵育20 min。DAB 辣根过氧化物酶显色后,采用苏木精染细胞核,随后乙醇梯度浓度脱水、二甲苯透明,封片后于显微镜下进行观察,统计CXCL9阳性、CD8α阳性的细胞比例以及染色强度,并进行病理评分。具体标准如下:① 对阳性细胞的占比进行统计,即小于5%记为0分,5%~25%记为1分,26%~50%记为2分,51%~75%记为3分,>75%记为4分。② 对阳性细胞的染色强度进行统计,即淡黄色记为1分,棕黄色记为2分,棕褐色记为3分。③ 将前述阳性细胞占比得分和染色强度得分相乘获得每个样本的分数,再将所有样本分数行Wilcoxon秩和检验。
1.5.2 乳腺癌间质CXCL9+ 细胞种类分析
采用多重荧光免疫组织化学染色(multiplex immunohistochemistry staining,mIHC)分析组织芯片中乳腺癌组织间质内的CXCL9+细胞种类,即将芯片进行烘烤、二甲苯脱蜡、乙醇梯度浓度水化、通用型强力抗原修复液水浴、自然冷却、内源性生物素封闭液封闭后,室温下一抗(CXCL9抗体的工作浓度为1∶200)孵育1~2 h或4 ℃孵育过夜,而后于室温下使用多聚辣根过氧化物酶标记的鼠兔混合二抗孵育10 min,荧光染料室温孵育10 min。
而后,在同一张芯片上重复3轮上述从抗原修复至荧光染料孵育的步骤,每轮仅孵育一种抗体(分别为CK7抗体、CD68抗体、CD11c抗体,工作浓度依次为1∶500、1∶500、1∶300),顺序自定,直至所有一抗被荧光染料逐一标记完成。于室温下,用DAPI工作液孵育芯片5 min,抗荧光淬灭封片剂进行封片。最后,采用全自动定量病理成像分析系统扫描组织芯片,观察乳腺癌组织间质内CXCL9阳性的细胞类型。
1.6 统计学分析
使用R语言软件和GraphPad Prism 9软件进行数据分析和可视化作图。使用Wilcoxon秩和检验进行2组间比较,使用Kruskal-Wallis检验进行3组及以上比较。使用KM生存曲线对CXCL9 mRNA表达或TIIC浸润水平与乳腺癌患者OS的相关性进行分析。P<0.05表示差异具有统计学意义。
2 结果
2.1 乳腺癌微环境中TIIC亚群的差异分析和对患者预后影响
利用CIBERSORT反卷积算法提供的22种TIIC基因表达特征集,对乳腺癌组织及癌旁组织的转录组数据进行免疫浸润细胞分析。图1A展示了乳腺癌组织和癌旁组织中22种TIIC亚群的占比情况的柱状图;聚类热图的结果(图1B)显示,乳腺癌组织和癌旁组织中各TIIC亚群间的分布比例具有明显差异。随后,我们对22种TIIC亚群在4种乳腺癌分子分型中的占比进行比较,结果(图1C)发现多数TIIC亚群在不同亚型中的占比差异明显,其中CD8+ T细胞、滤泡辅助T细胞、活化NK细胞、巨噬细胞M0、巨噬细胞M1在TN型和HER2+型中的比例明显高于Luminal型,活化的肥大细胞和巨噬细胞M2在Luminal型中的占比远高于另外2种分型(均P<0.05)。采用KM生存曲线分析TIIC浸润水平的高低与乳腺癌患者OS的相关性,结果(图2)显示在22种TIIC中,仅CD8+ T细胞、巨噬细胞M1、滤泡辅助T细胞和γδT细胞的高浸润与乳腺癌患者较长的OS相关(均P<0.05),巨噬细胞M2的高浸润则与患者较短的OS相关(P=0.000)。
图1
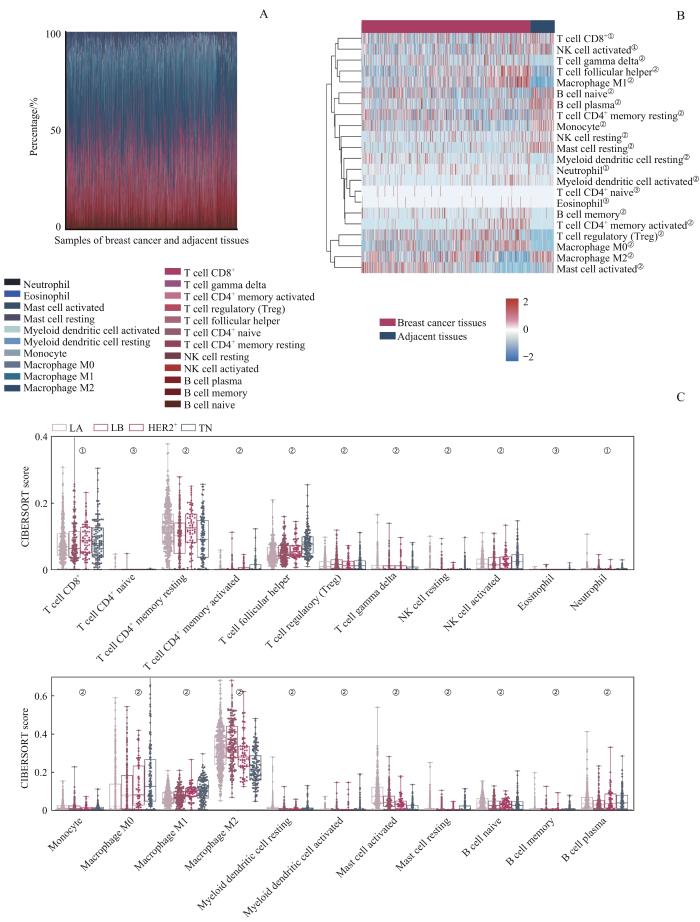
图1
乳腺癌TME中TIIC亚群的差异分析
Note: A. Proportions of 22 TIIC subgroups in the microenvironment of breast cancer and adjacent tissues. B. Comparison of the distributions of TIIC subgroups in breast cancer tissues and adjacent tissues by heatmap (①P=0.010, ②P=0.001, ③P>0.05). C. Differential analysis of 22 types of TIICs in different molecular types of breast cancer (①P=0.010, ②P=0.001, ③P>0.05).
Fig 1
Differential analysis of TIIC subgroups in breast cancer TME
图2
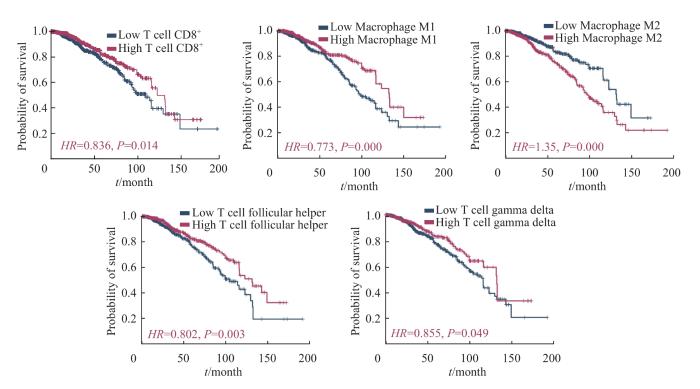
图2
TME中TIIC浸润水平对乳腺癌患者预后影响
Fig 2
Effect of TIICs infiltration ratio in TME on the prognosis of breast cancer patients
2.2 乳腺癌中目的基因的筛选及其表达水平与患者预后的相关性分析
采用R语言对来自TCGA数据库的乳腺癌组织及癌旁组织的转录组数据进行分析,绘制乳腺癌上调基因和下调基因火山图(图3A);其中,将log2(FC)>2的275个上调基因作为基因集一。从ImmPort数据库获取1 793个免疫相关基因作为基因集二。从GEPIA2数据平台筛选出影响乳腺癌患者生存的前500个基因作为基因集三。利用R语言分析上述3个基因集相交关系并绘制韦恩图,筛选获得目的基因——CXCL9(图3B)。随后,通过GEPIA数据平台分析CXCL9 mRNA在不同类型癌症中的表达水平,和部分癌种及其对应正常组织的差异表达,结果(图4A)显示CXCL9 mRNA在乳腺癌、膀胱尿路上皮癌、胆管细胞癌、结肠腺癌、食管癌、胶质母细胞瘤、头颈部鳞状细胞癌、肾透明细胞癌、肝细胞癌、肺腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、胃腺癌、子宫内膜癌等癌组织中的表达均高于其对应的正常组织(均P<0.05)。
图3
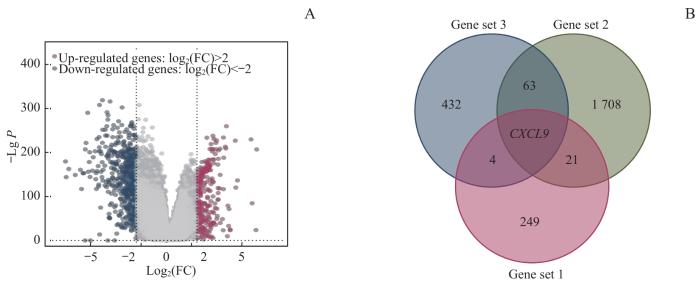
图3
目的基因的筛选
Note: A. Volcano plot of DEGs between breast cancer tissues and adjacent tissues. B. Venn diagram of the target gene screened in the three gene sets.
Fig 3
Screening of target gene
图4
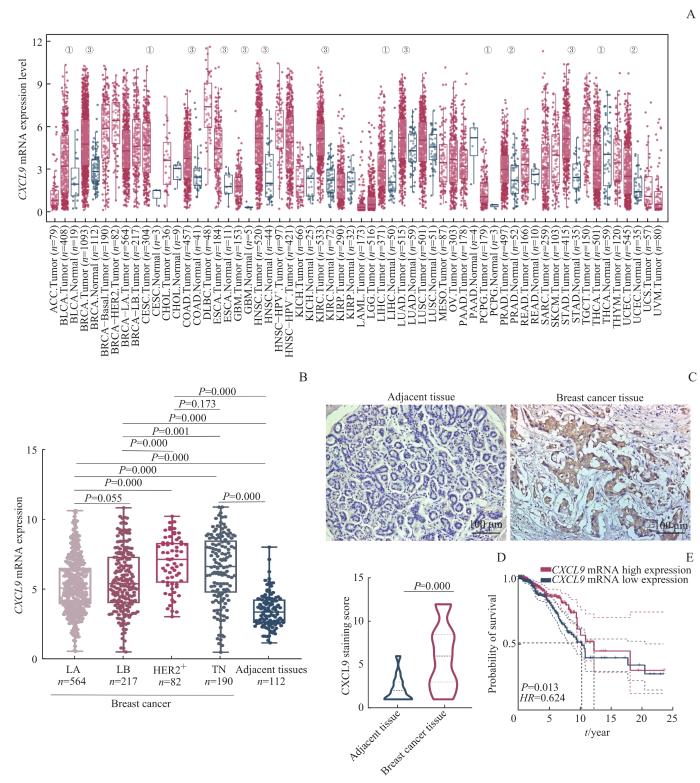
图4
目的基因的表达水平对乳腺癌患者的预后影响
Note: A. Expression of CXCL9 mRNA in different types of tumor and their corresponding normal tissues (①P=0.050, ②P=0.010, ③P=0.001). ACC—adrenocortical carcinoma; BLCA—bladder urothelial carcinoma; BRCA—breast invasive carcinoma; CESC—cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL—cholangiocarcinoma; COAD—colon adenocarcinoma; DLBC—lymphoid neoplasm diffuse large B-cell lymphoma; ESCA—esophageal carcinoma; GBM—glioblastoma multiforme; HNSC—head and neck squamous cell carcinoma; KICH—kidney chromophobe; KIRC—kidney renal clear cell carcinoma; KIRP—kidney renal papillary cell carcinoma; LAML—acute myeloid leukemia; LGG—brain lower grade glioma; LIHC—liver hepatocellular carcinoma; LUAD—lung adenocarcinoma; LUSC—lung squamous cell carcinoma; MESO—mesothelioma; OV—ovarian serous cystadenocarcinoma; PAAD—pancreatic adenocarcinoma; PCPG—pheochromocytoma and paraganglioma; PRAD—prostate adenocarcinoma; READ—rectum adenocarcinoma; SARC—sarcoma; SKCM—skin cutaneous melanoma; STAD—stomach adenocarcinoma; TGCT—testicular germ cell tumor; THCA—thyroid carcinoma; THYM—thymoma; UCEC—uterine corpus endometrial carcinoma; UCS—uterine carcinosarcoma; UVM—uveal melanoma. B. Boxplot of CXCL9 mRNA expression in four molecular types of breast cancer and adjacent tissues. C/D. Observation (C) and statistical analysis (D) of differential expression of CXCL9 in breast cancer tissues and adjacent tissues by IHC. E. Detection of the effect of CXCL9 mRNA expression on prognosis of breast cancer patients by KM survival curve.
Fig 4
Effect of the expression of target genes on the prognosis of breast cancer patients
采用Kruskal-Wallis检验对乳腺癌(不同分子分型)及其癌旁组织中CXCL9 mRNA的表达水平进行分析,结果(图4B)显示,CXCL9 mRNA在乳腺癌的4种分子分型中的表达水平高于癌旁组织(均P=0.000),在TN型和HER2+ 型中的表达高于Luminal型(均P<0.05)。利用IHC对组织芯片进行分析,结果(图4C、D)显示相较于癌旁组织,乳腺癌组织内肿瘤上皮细胞普遍高表达CXCL9,且差异具有统计学意义(P=0.000),与上述生物数据库的分析结果一致。为进一步探讨CXCL9 mRNA表达与患者生存之间的关系,我们使用KM生存曲线评估CXCL9 mRNA高表达和低表达对患者预后的影响。结果(图4E)显示,CXCL9 mRNA高表达与乳腺癌患者较长的OS有关(P=0.013)。
2.3 CXCL9的PPI网络构建和 CXCL9 及其相关基因的GO功能、KEGG通路分析
为进一步探讨CXCL9基因在肿瘤发生中的分子机制,本研究对CXCL9相关的PPI网络蛋白对应的基因、乳腺癌中与CXCL9表达正相关的基因行GO功能分析和KEGG富集分析。图5A显示构建的PPI网络包括了41个蛋白,我们将其对应的基因记为数据集一。同时,将乳腺癌中与CXCL9表达正相关的前150位基因记为数据集二。将上述2个数据集合并,运用R语言对其行GO功能分析和KEGG通路分析。GO功能分析的结果(图5B)显示,多数基因与免疫细胞的生物过程有关,如适应性免疫应答、调节T细胞活化、免疫系统过程的负调控、免疫效应过程的调节、白细胞分化的调节、细胞因子产生的正调控等。KEGG通路分析的结果(图5C)显示,CXCL9可能通过参与病毒蛋白与细胞因子或细胞因子受体的相互作用、T细胞受体信号传导、自然杀伤细胞介导的细胞毒性等通路对TME中TIIC进行调节,来影响肿瘤发展及患者预后。
图5
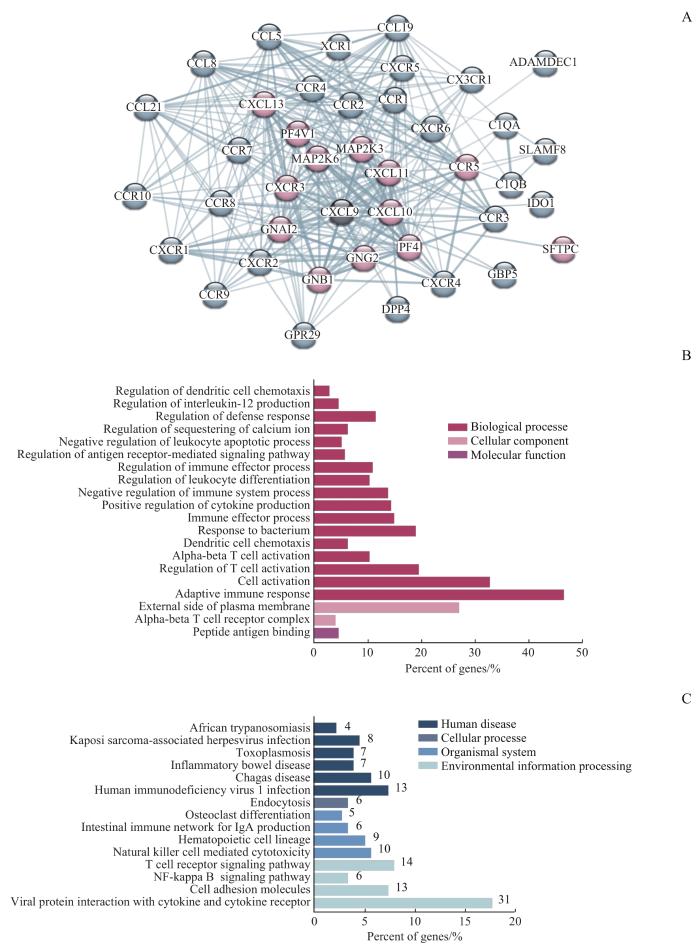
图5
CXCL9的PPI网络构建和 CXCL9 及其相关基因的GO功能分析和KEGG通路分析
Note: A. PPI network of CXCL9. Red indicates proteins with CXCL9 interaction score >0.9, and blue indicates proteins with CXCL9 interaction score >0.7. PF4—platelet factor 4; PF4V1—platelet factor 4 variant 1; MAP2K3/6—dual specificity mitogen-activated protein kinase kinase 3/6; GNAI2—guanine nucleotide-binding protein G(i) subunit alpha-2; GNG2—guanine nucleotide-binding protein G(I)/G(S)/G(O) subunit gamma-2; GNB1—guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1; SFTPC—surfactant protein C; CCR1—C-C chemokine receptor type 1; CXCR1—C-X-C chemokine receptor type 1; CX3CR1—CX3C chemokine receptor 1; CCL5—C-C motif chemokine ligand 5; DPP4—dipeptidyl peptidase-4; GPR29—chemokine (C-C motif) receptor 6; XCR1—chemokine XC receptor 1; C1QA/B—complement component 1, Q subcomponent, A chain/B chain; IDO1—indoleamine 2,3-dioxygenase 1; SLAMF8—SLAM family member 8; ADAMDEC1—adam-like, decysin 1; GBP5—guanylate-binding protein 5. B/C. GO function analysis (B) and KEGG pathway analysis (C) of CXCL9 and its related genes.
Fig 5
PPI network construction of CXCL9 and GO functional analysis and KEGG pathway analysis of CXCL9 and its related genes
2.4 乳腺癌中 CXCL9mRNA表达与TIIC相关性分析
本研究进一步探讨乳腺癌中CXCL9 mRNA表达水平与TIIC浸润丰度的关系。采用Spearman相关系数分析乳腺癌微环境中免疫细胞浸润评分与CXCL9 mRNA表达的相关性并绘制热图。结果(图6A)显示,CXCL9 mRNA表达水平主要与记忆B细胞、CD8+ T细胞、CD4+ 记忆T细胞、γδT细胞、滤泡辅助T细胞、巨噬细胞M1等TIIC的浸润比例呈正相关(均P<0.05),与静息型自然杀伤细胞、巨噬细胞M0、巨噬细胞M2、活化髓系树突状细胞、肥大细胞等的浸润比例呈负相关(均P<0.05)。
图6
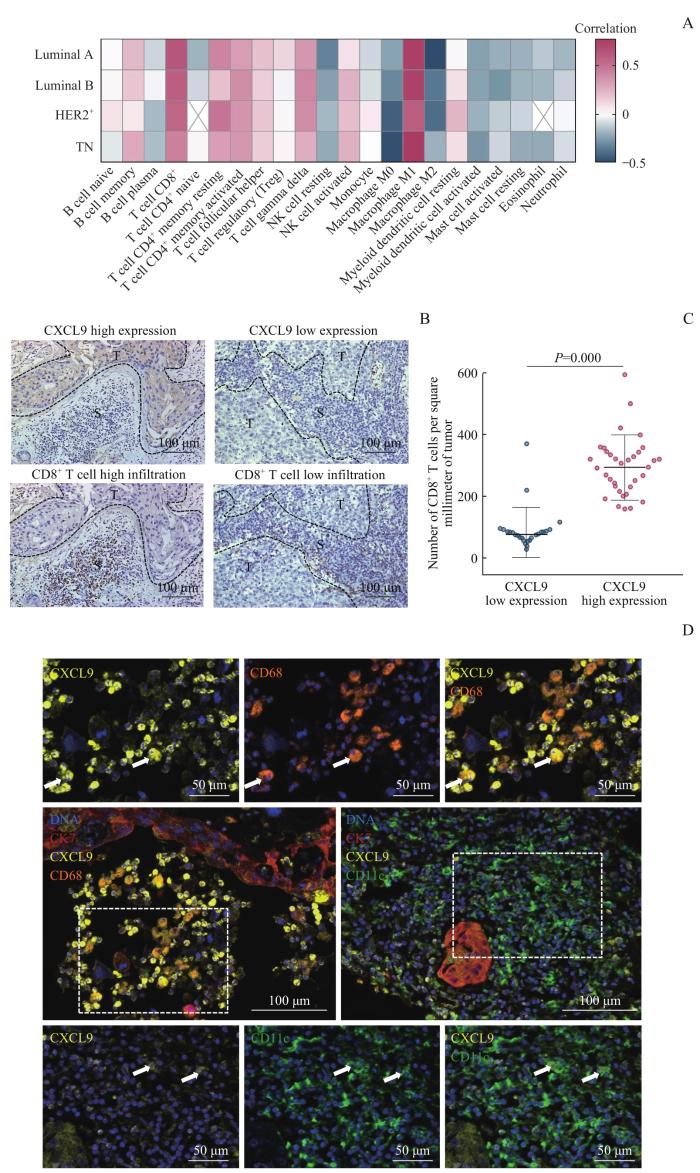
图6
乳腺癌中 CXCL9 mRNA表达与TIIC浸润水平的相关性分析
Note: A. Spearman correlation analysis of TIICs infiltration ratio and CXCL9 mRNA expression. B. Observation of the relationship between CXCL9 expression and CD8+ T cells infiltration ratio by IHC. S—stroma; T—tumor. C. Correlation between CXCL9 expression and the number of CD8+ T cells infiltration by beeswarm plot. D. Fluorescence images of CXCL9+ cell species in breast cancer interstitium. Yellow indicates CXCL9, orange indicates CD68, green indicates CD11c, red indicates CK7, and blue indicates DNA. The left dashed box indicates the field of upper 3 small figures, and the right one indicates the field of lower 3 small figures. The arrows indicate CXCL9+CD68+ cells or CXCL9+CD11c+ cells.
Fig 6
Correlation between CXCL9 mRNA expression and TIICs infiltration ratio in breast cancer
2.5 乳腺癌中 CXCL9mRNA表达水平和CD8+ T细胞浸润高低与患者预后的分析
本研究使用Spearman相关系数分析CXCL9 mRNA表达和TIIC浸润比例、免疫检查点基因表达水平之间的相关性,以探讨CXCL9在乳腺癌微环境中相关的免疫调控机制。结果(图7A、B)显示,CXCL9 mRNA与大多数的免疫检查点基因呈强正相关性;CD8+ T细胞浸润比例与多数免疫检查点基因的表达亦呈正相关(均P<0.05),其中LAG3(Rho=0.648)、PDCD1(Rho=0.809)、CTLA4(Rho=0.723)、TIGIT(Rho=0.821)这4个基因和CD8+ T细胞的正相关性更为明显。为进一步探究CXCL9招募CD8+ T细胞的潜在作用及其影响患者预后的机制,我们对两者开展KM生存曲线分析。结果(图7C)显示,CXCL9 mRNA低表达时,CD8+ T细胞高浸润、低浸润的乳腺癌患者预后间差异无统计学意义;而CXCL9高表达时,与CD8+ T细胞低浸润相比,细胞高浸润与乳腺癌患者的良好预后相关(P=0.001)。
图7
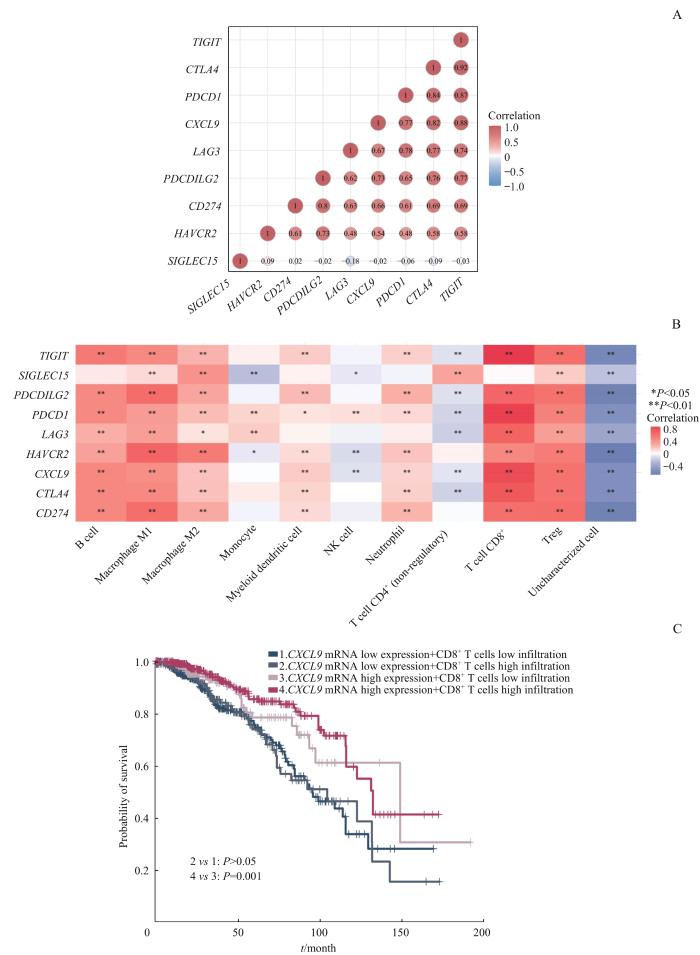
图7
CXCL9 mRNA表达水平和CD8+ T细胞浸润比例对乳腺癌患者预后的影响
Note: A. Correlation between CXCL9 mRNA and immune checkpoint genes. B. Heatmap of various TIICs infiltration ratio in relation to CXCL9 and immune checkpoint genes. C. Effect of combination of high and low expression of CXCL9 mRNA and high and low infiltration of CD8+ T cells on the prognosis of breast cancer patients.
Fig 7
Effect of CXCL9 mRNA expression level and CD8+ T cells infiltration ratio on the prognosis of breast cancer patients
3 讨论
本研究通过生物信息学数据库分析,描述了乳腺癌微环境的特征,筛选出目的基因CXCL9;且相较于癌旁组织,CXCL9 mRNA在乳腺癌中表达更高,KM生存曲线显示其高表达与乳腺癌患者较长的OS相关。同时,先前的研究[26]亦证明,在小鼠乳腺癌模型中CXCL9过表达组表现出更好的预后。继而提示,CXCL9或可作为预测乳腺癌预后的生物因子。而后,我们通过GO功能和KEGG通路分析发现,CXCL9及其相关基因主要在免疫调控相关功能和通路富集;分析目的基因的mRNA水平与TIIC的相关性后发现,CXCL9 mRNA与部分TIIC浸润比例呈正相关。IHC实验的结果显示,乳腺癌中CXCL9的表达与CD8+ T细胞浸润数目呈正相关;KM生存曲线分析发现,只有在CXCL9 mRNA高表达时,CD8+ T细胞高浸润才能延长乳腺癌患者的生存期。继而我们推测,CXCL9在乳腺癌微环境中可能是通过调控部分TIIC的迁移、激活等功能使患者获得更长的OS。YU等[27]分析结直肠癌微环境中的TIIC后发现,CXCL9~11 mRNA与自然杀伤细胞、CD8+ T细胞的浸润比例高度相关;亦有小鼠模型的研究[28-29]证明CXCL9可通过招募自然杀伤细胞、激活T细胞来减少肿瘤生长和转移。继而提示,CXCL9招募TIIC后可能会进一步调控TIIC的抗肿瘤作用,从而使患者获得更好的预后。此外,通过mIHC实验发现,乳腺癌间质中部分CD68+ TAM和CD11c+ DC均可表达CXCL9。同样,最近报道[30]显示CXCL9在卵巢TME中的表达仅限于TAM和DC中,且CXCL9的表达依赖于肿瘤抗原的识别并由干扰素-γ(interferon-γ,IFN-γ)特异性诱导上调。分析上述调控的原因,其可能的解释是前馈循环假说,即新招募的T细胞在抗原参与下产生IFN-γ以诱导TAM和DC产生额外的CXCL9,进而可招募更多的T细胞进入TME,从而形成“热”肿瘤微环境,但该解释尚需进一步的功能实验研究验证。
本研究通过生物信息学、组织标本验证等手段,初步证明CXCL9可能可以作为评估乳腺癌患者预后和微环境免疫浸润类型的生物学标志物;并初步揭示了CXCL9可能是通过招募部分TIIC浸润并激活CD8+ T细胞抗肿瘤功能抑制乳腺癌进展,从而延长患者的生存期。该结果或将为CXCL9作为乳腺癌的良好预后标志物和治疗靶点提供新的证据。在今后的工作中,我们将继续探索CXCL9调节乳腺癌免疫微环境的分子效应及促进CD8+ T细胞发挥抗肿瘤作用的分子机制,从而优化乳腺癌患者的临床免疫治疗策略。
作者贡献声明
臧丽娟、范广建、王红霞和胡孝渠参与了本研究的设计,杜少倩完成了生物信息学分析、临床组织验证实验及文章撰写,陶梦玉参与部分内容撰写,曹源参与部分生物信息学分析。所有作者均阅读并同意了最终稿件的提交。
ZANG Lijuan, FAN Guangjian, WANG Hongxia and HU Xiaoqu designed the study. DU Shaoqian completed bioinformatics analysis, clinical tissue validation experiments and article writing. TAO Mengyu participated in part of article writing. CAO Yuan participated in part of bioinformatics analysis. All the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
All authors disclose no relevant conflict of interests.
参考文献

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}