

近年来,肿瘤研究日益关注肿瘤微环境(tumor microenvironment,TME)的重要作用。TME由多种细胞类型构成,包括免疫细胞、内皮细胞和基质细胞等。而肿瘤相关巨噬细胞(tumor-associated macrophages,TAMs)被认为是TME的主要组成部分,可分为具有抗肿瘤活性的M1巨噬细胞和促进肿瘤发生发展的M2巨噬细胞。现有研究[3-5]表明,TAMs的高浸润与多数恶性肿瘤的不良预后与治疗耐药性密切相关。有学者[6]指出,去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)中的TAMs会积累胆固醇并将其转移至癌细胞中,这些胆固醇可作为雄激素生物合成的前体,激活癌细胞内的雄激素受体(androgen receptor,AR)。研究[7]表明,雄激素剥夺治疗时TAMs会过度表达和分泌白细胞介素-1β(interleukin-1β,IL-1β),IL-1β进一步诱导骨髓来源抑制细胞的积累,并抑制细胞毒性T细胞的激活,从而形成免疫抑制性微环境。近年来,普通转录组测序(RNA sequencing,RNA-seq)和单细胞转录组测序(single cell RNA sequencing,scRNA-seq)技术迅速发展,作为互补的转录组分析手段,它们从不同层面揭示了TME在PCa进展及预后预测中的作用。
当前,已有多个基于临床变量和基因组数据的PCa预后模型被提出,旨在提高预测准确性与临床决策的可靠性。这些预后模型,如格里森评分(Gleason Score,GS)、美国癌症联合委员会(American Joint Committee on Cancer,AJCC)分期、前列腺癌风险评估(Cancer of the Prostate Risk Assessment,CAPRA)评分等,虽然在临床上广泛应用,但它们通常依赖于临床和病理数据,未能充分考虑肿瘤分子特征的影响,因此存在一定局限性。此外,现有的基于肿瘤基因组数据的模型,如Prolaris检测、Oncotype DX检测等,虽然有一定优势,但由于样本量有限且未考虑到TME对肿瘤发展的影响,仍存在一定的不确定性和应用局限性[8-10]。鉴于M2巨噬细胞在TME中促肿瘤的关键作用,本研究结合RNA-seq和scRNA-seq进行综合分析,并基于M2巨噬细胞基因特征构建更为可靠的PCa预后预测模型。
1 资料与方法
1.1 数据获取与处理
1.2 免疫浸润与生存分析
1.3 单细胞分析
使用Seurat包生成单细胞分析对象,对每个样本进行质量控制。使用harmony包整合Seurat对象以消除批次效应,并将每个细胞投影到tSNE图中。使用FindMarkers函数识别scRNA-seq数据中的差异基因,并用AddModuleScore函数对scRNA-seq数据进行评分。采用enrichGO函数进行基因本体(Gene Ontology,GO)富集分析,筛选条件为FoldChange>1.5和调整后的P值<0.05[17]。使用GSEA函数进行基因集富集分析(Gene Set Enrichment Analysis,GSEA),所用基因集来自MSigDB数据库。通过CellChat算法分析细胞间通信情况[18]。
1.4 风险评分模型的建立
使用单因素Cox回归分析筛选预后相关基因,将P<0.05的基因纳入LASSO回归分析,并通过glmnet包进行LASSO分析[19]。基于M2巨噬细胞特征构建风险评分模型,其公式为:RiskScore=基因1×β1+基因2×β2+基因3×β3+……+基因n×βn。使用maxstat包确定最大对数秩统计量对风险评分进行二分法分析。通过timeROC包绘制受试者工作特征(receiver operating characteristic,ROC)曲线,并计算曲线下面积(area under curve,AUC)和一致性指数(concordance index,C-index)评估该预后模型的预测准确性。基于预后特征与样本临床信息,使用rms包构建列线图。
1.5 统计学分析
所有统计分析均在R软件(版本4.1.1)中进行。使用Wilcoxon检验分析高风险组和低风险组之间的免疫浸润与免疫检查点表达情况差异。使用Kaplan-Meier进行生存分析。P<0.05表示差异具有统计学意义。
1.6 化疗药物敏感性预测
使用oncoPredict软件预测高风险组与低风险组患者对不同药物的半抑制浓度(half maximal inhibitory concentration,IC50),并通过Spearman相关系数评估IC50与风险评分之间的相关性。采用Wilcoxon秩和检验筛选高风险组与低风险组之间差异最显著的前12种药物。
2 结果
2.1 M2巨噬细胞高度浸润的PCa患者预后不良
首先通过CIBERSORTx算法评估TCGA患者样本的免疫浸润情况。生存分析结果显示,M2巨噬细胞高浸润患者的无进展生存期(progression-free survival,PFS)显著缩短(P<0.001),而M0与M1巨噬细胞浸润状态与患者预后无关(P=0.830,P=0.700,图1A~C)。
图1
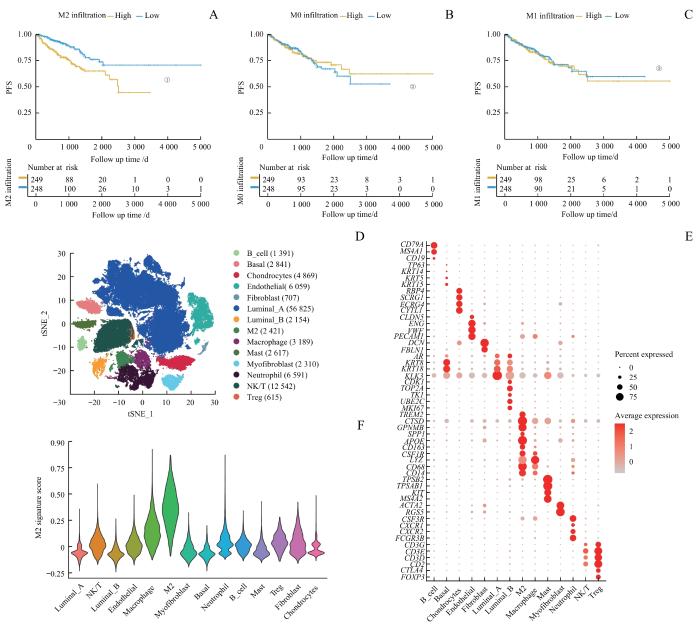
图1
PCa中M2巨噬细胞相关特征的鉴定
Note: A‒C.Comparison of PFS between low and high infiltration groups for M2 (A), M0 (B) and M1 (C) macrophages. D. Dimension reduction plot displaying the composition of 14 major cell clusters derived from prostate cancer samples. E. Bubble plot showing the marker genes of each cell cluster. F. Violin plot presenting the M2 signature scores across each cell cluster. ①P<0.001, ②P=0.830, ③P=0.700.
Fig 1
Identification of M2 macrophage-related characteristics in PCa
2.2 M2巨噬细胞在TME中发挥促肿瘤作用
对scRNA-seq数据集进行质量控制后,纳入105 131个细胞。通过表面标志物对细胞进行分类,得到14种细胞类型,包括基底细胞(2 841个)、管腔上皮A型细胞(56 825个)、管腔上皮B型细胞(2 154个)、内皮细胞(6 059个)、成纤维细胞(707个)、肌成纤维细胞(2 310个)、软骨细胞(4 869个)、B细胞(1 391个)、M2巨噬细胞(2 421个)、非M2巨噬细胞(2 617个)、单核细胞(3 189个)、中性粒细胞(6 591个)、Treg细胞(615个)和NK/T细胞(12 542个),计算每种细胞类型的M2特征基因评分(图1D~F)。差异基因分析显示:M2巨噬细胞高表达APOE、GPNMB、SPP1、C1QC等基因,主要参与MHC Ⅱ类分子介导的抗原提呈和免疫球蛋白介导的免疫反应;而非M2巨噬细胞则高表达IL1B、S100A8、S100A9等基因,主要参与淋巴细胞激活、黏附与吞噬作用(图2A、B)。
图2
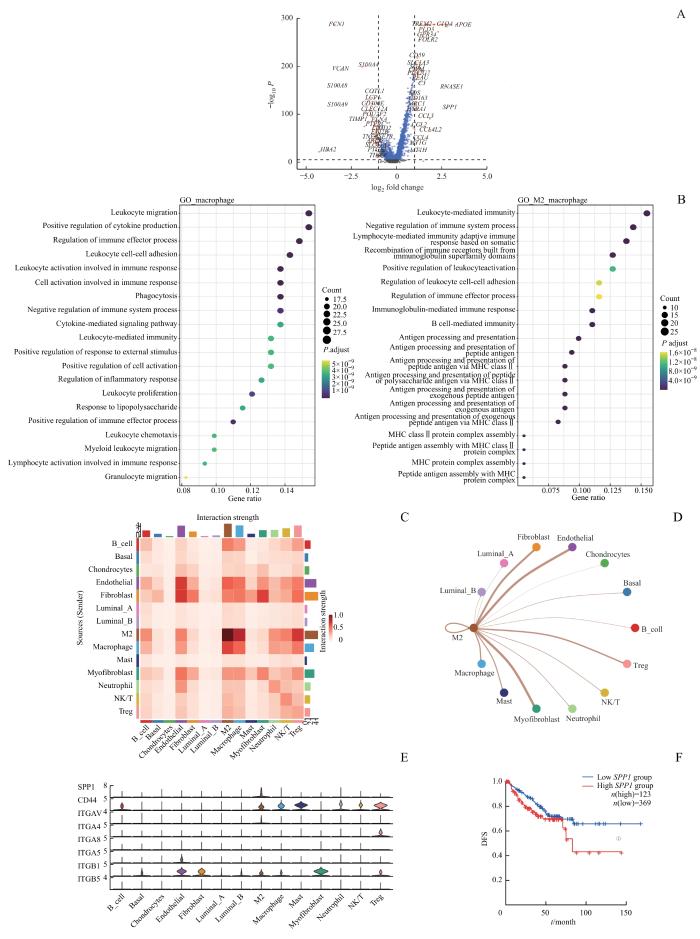
图2
TME中细胞间相互作用分析
Note: A. Volcano plot illustrating the differentially expressed genes between M2 macrophages and non-M2 macrophages (genes with significant differences are highlighted in red; non-significant genes in blue). B. Gene enrichment analysis represented as a bubble diagram. C/D. Heatmap (C) and network plot (D)visualizing cell communication in sc-RNA seq. E. Violin plot indicating the predominant secretion of SPP1 by M2 macrophages. F. Kaplan-Meier survival curve showing DFS difference between high and low SPP1 expression groups in the TCGA cohort. ①P=0.034.
Fig 2
Analysis of cell-cell interactions in TME
细胞通信分析表明,与非M2巨噬细胞相比,M2巨噬细胞与调节性T细胞(Treg细胞)、NK/T细胞和中性粒细胞的相互作用更强(图2C、D)。M2巨噬细胞可以分泌SPP1,广泛影响TME中多种免疫细胞,包括Treg细胞、NK/T细胞、中性粒细胞、B细胞和肥大细胞(图2E)。在TCGA队列中,高表达SPP1的患者也显著表现出更短的DFS(P=0.034,图2F)。而最新研究[20]也表明,晚期PCa患者中的TAMs分泌SPP1从而导致免疫抑制。此外,M2巨噬细胞还可以表达较高水平的CCL3、CCL4等,它们在之前研究中被认为与PCa的生化复发与骨转移相关[21-24]。细胞通信分析显示,M2巨噬细胞可以通过表达CCL3、CCL4,作用于TME中的NK/T细胞、Treg细胞和B细胞,进而促进肿瘤免疫抑制性微环境的形成,影响疾病进展(图3A~C)。同时,TCGA数据表明,患者表达CCL3和CCL4的水平与DFS显著相关(P=0.036,P=0.032,P=0.013,图3D~F)。这些证据进一步验证了M2巨噬细胞在PCa TME中促肿瘤的关键作用。
图3
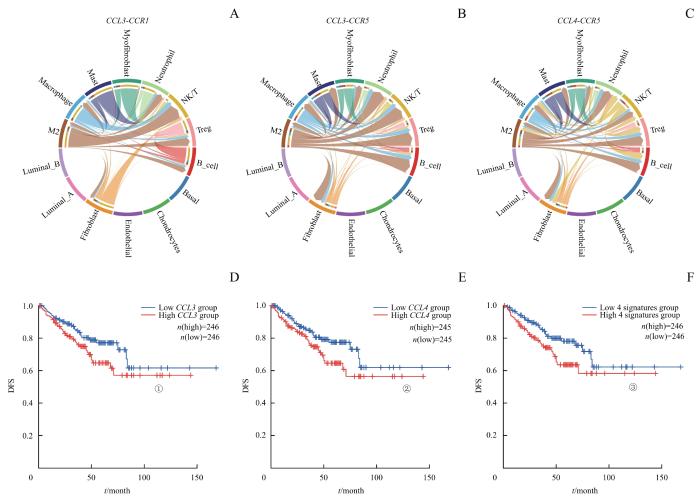
图3
M2巨噬细胞与TME中其他细胞交互的主要通路
Note: A‒C. The signals of CCL3 and CCL4 from M2 macrophages primarily targeted NK/T cells, Treg cells and B cells in PCa. D‒F. DFS differences based on these pathways. ①P=0.036, ②P=0.032, ③P=0.013.
Fig 3
Major pathways of interaction between M2 macrophages and other cells in TME
2.3 PCa风险评分预后模型的构建和验证
基于M2巨噬细胞在TME中的关键作用及其与肿瘤不良预后的关联,利用M2巨噬细胞特征基因构建一种PCa风险评分模型,以预测患者预后。首先,从scRNA-seq数据集中识别出108个与M2巨噬细胞浸润显著相关的基因(Spearman相关系数>0.3,P<0.05)以及554个在M2 TAMs中高表达的基因,并将这些基因集取交集。将TCGA队列中的497例非转移性PCa患者按6∶4的比例随机分为298例训练集和199例测试集。在训练集中,基于单因素Cox回归分析得到38个与PFS显著相关的基因。基于这38个基因构建LASSO回归模型,并筛选出具有最低交叉验证误差的4个特征基因:TREM2、OTOA、SIGLEC1和PLXDC1(图4A~C)。风险评分模型公式为:RiskScore=0.095 257 59×TREM2+0.081 748 26×OTOA+0.097 203 08×SIGLEC1+0.171 846 15×PLXDC1。
图4
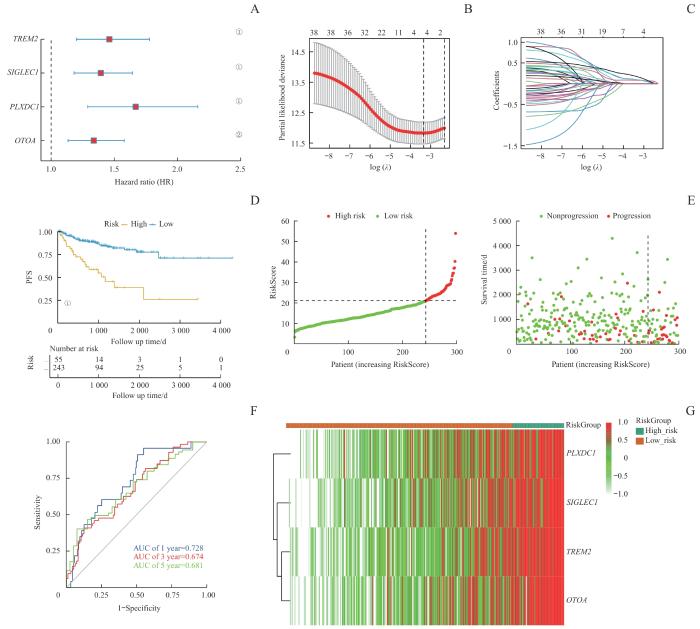
图4
PCa预后模型的构建与测试
Note: A. The construction of a LASSO regression model based on four characteristic genes (TREM2, OTOA, SIGLEC1, and PLXDC1). B. Identification process of genes for construction of prognostic risk score signature. C. The coefficients in LASSO Cox regression analysis of the 38 prognostic M2 macrophages-related genes. D. The comparison of PFS between low- and high-risk score groups in the training set. E. The distribution of risk scores ranked from low to high and the comparison of survival status between low- and high- risk score groups in the training set. F. The prediction accuracy of the risk score measured by ROC curves at 1, 3, 5 years in the training set. The AUC values are 0.728, 0.674, 0.681 respectively. ①P<0.001, ②P=0.001.
Fig 4
Construction and testing of the prognostic model in PCa
研究表明,在训练集中,相比于低风险评分的患者,高风险评分的患者具有更低的PFS,这突显了M2巨噬细胞对PCa复发的重要作用(P<0.001,图4D~F)。使用ROC曲线评估模型性能,结果显示1、3和5年的AUC分别为0.728、0.674和0.681(图4G)。随后,在测试集中验证了该风险评分模型,结果类似,高风险评分的患者表现出较低的PFS(P=0.018,图5A)。在整个TCGA队列中,1、3和5年的AUC值分别为0.693、0.659和0.649,表明该模型具有良好的预测性能(图5B~D)。此外,本研究还使用了2个独立的外部验证数据集,以进一步证实模型的可靠性。在MSKCC队列中,高风险评分组的患者显示出较高的生化复发(biochemical recurrence,BCR)风险(P<0.001,图5E)。而对于SU2C/PCF Dream Team队列中的转移性PCa患者,高风险评分组个体也表现出较低的总生存期(overall survival,OS)(P=0.013,图5F)。
图5
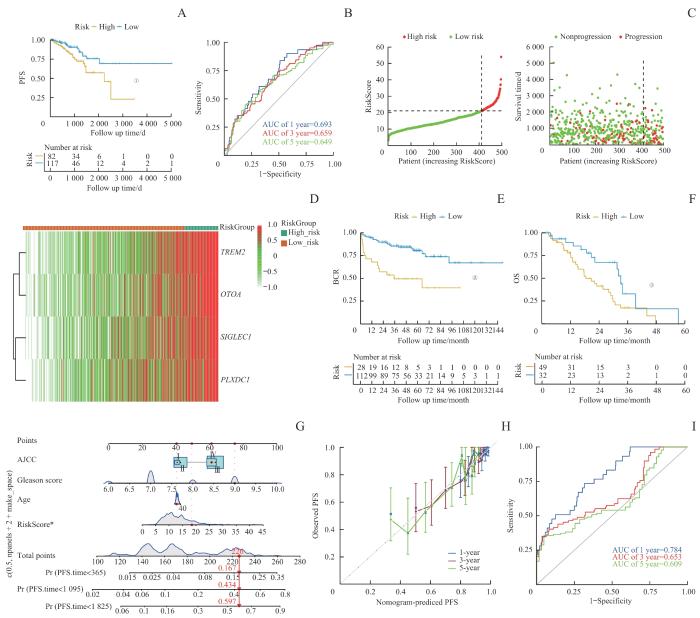
图5
通过外部数据库对预后模型进行验证
Note: A. The comparison of PFS between low- and high-risk score groups in the test set. B. The prediction accuracy of the risk score measured by ROC curves at 1, 3, 5 years in the test set. The AUC values were 0.693, 0.659, and 0.649 for 1, 3, and 5 years, respectively. C. Distribution of risk in ascending order and comparison of survival status between low- and high- risk score groups in the test set. D. The heatmap of four signature genes between high- and low-risk group. E/F. The comparison of BCR (E) and OS (F) between low- and high-risk score groups in the SU2C/PCF Dream Team cohort. G. Nomogram for predicting PFS of patients in training set. H. The calibration plots of the nomogram at 1, 3, 5 years. The x coordinate value represents the nomogram-predicted survival, and the y coordinate value represents observed PFS. I. ROC curves for nomogram at 1, 3, 5 years. ①P=0.018, ②P<0.001, ③P=0.013.
Fig 5
External database validation of prognostic model
表1 包含风险评分的多变量Cox回归模型
Tab 1
| Item | Coef | Exp(coef) | Se(coef) | Z | Pr(>|z|) |
|---|---|---|---|---|---|
| RiskScore | 0.041 055 | 1.041 909 | 0.020 700 | 1.983 | 0.047 3 |
| Age | -0.002 394 | 0.997 609 | 0.016 200 | -0.148 | 0.882 5 |
| Gleason score | 0.641 375 | 1.899 091 | 0.133 834 | 4.792 | 1.65×10-6 |
| AJCCⅡ | 0.050 297 | 1.051 583 | 1.060 082 | 0.047 | 0.962 2 |
| AJCCⅢ | 0.581 300 | 1.788 361 | 1.060 250 | 0.548 | 0.583 5 |
| AJCCⅣ | 0.524 520 | 1.689 648 | 1.087 386 | 0.482 | 0.629 5 |
表2 不包含风险评分的多变量Cox回归模型
Tab 2
| Item | Coef | Exp(coef) | Se(coef) | Z | Pr(>|z|) |
|---|---|---|---|---|---|
| Age | 0.000 538 3 | 1.000 538 5 | 0.015 995 0 | 0.034 | 0.973 |
| Gleason score | 0.705 826 4 | 2.025 519 8 | 0.131 378 0 | 5.372 | 7.77×10-6 |
| AJCCⅡ | -0.088 018 0 | 0.915 744 4 | 1.057 486 1 | -0.083 | 0.934 |
| AJCCⅢ | 0.488 412 6 | 1.629 727 2 | 1.059 586 6 | 0.461 | 0.645 |
| AJCCⅣ | 0.420 683 5 | 1.523 002 2 | 1.088 112 2 | 0.387 | 0.699 |
2.4 风险评分与患者临床特征的关联
图6
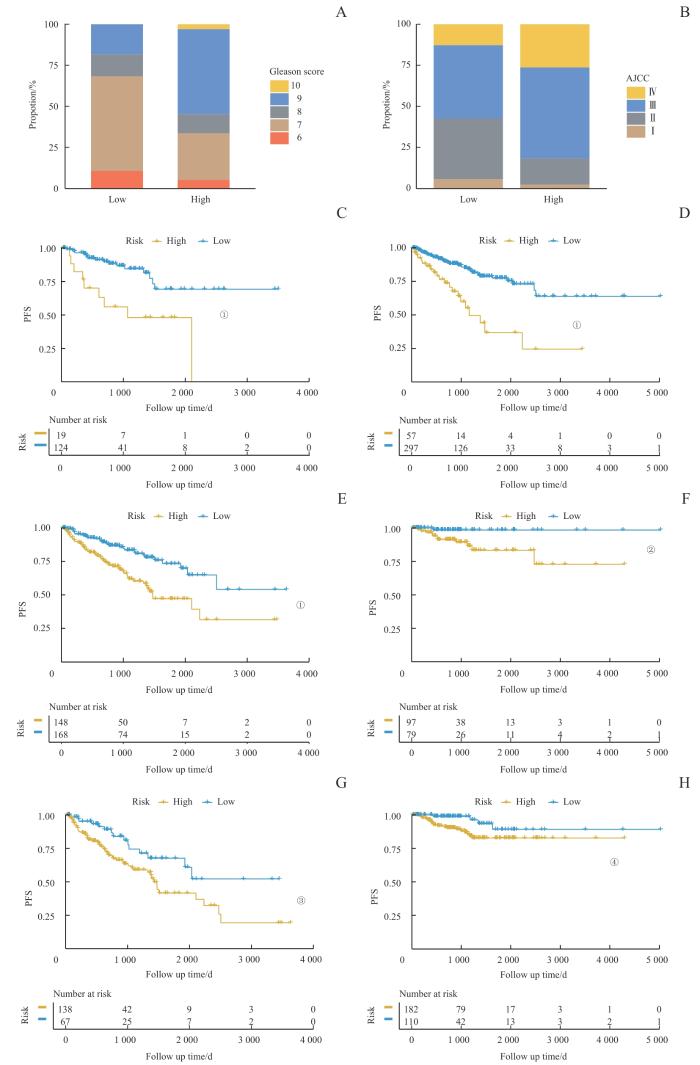
图6
临床特征与风险评分的关联
Note: A/B. The analysis of the RiskScore distribution based on Gleason score (A) and AJCC stage (B). C‒H. Kaplan-Meier analysis of PFS between high-risk and low-risk score groups among patients with different clinical pathological features. C. Patients with Gleason scores 6‒7. D. Patients with Gleason scores 8‒10. E. Patients with AJCC stage Ⅰ/Ⅱ. F. Patients with AJCC stage Ⅲ/Ⅳ. G. Patients aged ≤65 years. H. Patients aged >65 years. ①P<0.001, ②P=0.011, ③P=0.010, ④P=0.024.
Fig 6
Association of clinical characteristics with RiskScore
2.5 风险评分与免疫抑制与耐药性的关联
对TCGA队列数据中的高风险评分组和低风险评分组患者间的差异表达基因进行GSEA分析。结果显示:高风险评分组中高表达的基因显著富集于多个免疫抑制通路,包括血管生成通路、上皮-间质转化通路、IL-10信号通路、PD-1通路和CTLA-4通路(图7A、B)。对比免疫细胞浸润情况发现,高风险评分组中M2巨噬细胞和Treg细胞的浸润水平较高,而NK细胞和浆细胞的浸润水平则较低(图7C)。同时,高风险评分组的免疫检查点相关基因的表达水平显著提高,且不仅限于PDCD1、CD274和CTLA4等常见的免疫检查点基因(图7D)。综上,根据建立的预测模型计算出风险评分,能够成功将患者分为免疫抑制微环境组和非免疫抑制微环境组。
图7
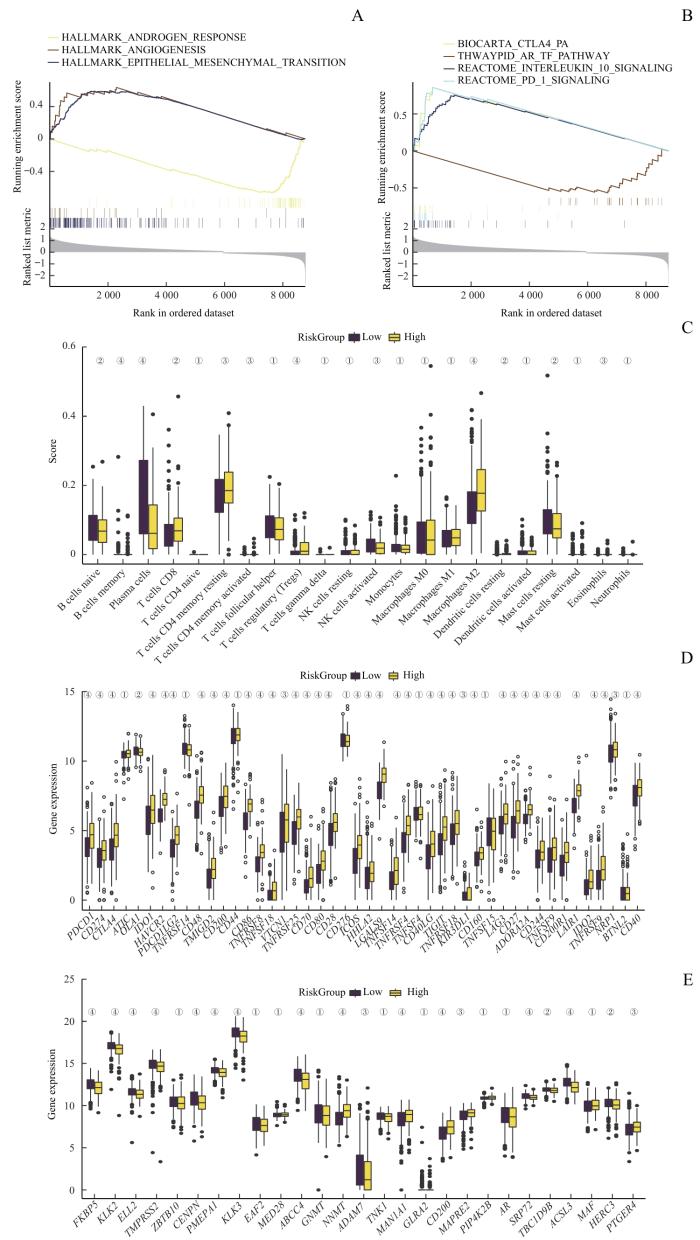
图7
风险评分与免疫抑制和耐药性的关联
Note: A/B. GSEA of patients with high or low Risk Score. C. Analysis of immune cell infiltration in PCa samples with high or low Risk Score. D. Analysis of immune checkpoint-related genes in PCa samples with high or low Risk Score. E. Analysis of
Fig 7
Association of immunosuppression and drug resistance with RiskScore
同时,低风险评分组的患者不仅拥有非免疫抑制性微环境,其AR通路活性也显著高于高风险评分组(图7E)。在临床上,内分泌治疗是PCa的一线治疗方法,它通过抑制AR通路活性来抑制癌细胞的生长。因此,考虑到低风险评分组患者中AR通路具有较高活性,可以推测这类患者可能对内分泌治疗更为敏感。
2.6 药物预测与敏感性分析
表3 与风险评分呈负相关的药物
Tab 3
| Drug ID | Drug name | Pathway name | Target |
|---|---|---|---|
| 1021 | Axitinib | RTK signaling | PDGFR, KIT, VEGFR |
| 1022 | AZD7762 | Cell cycle | CHEK1, CHEK2 |
| 1059 | AZD8055 | PI3K/mTOR signaling | MTORC1, MTORC2 |
| 1192 | GSK269962A | Cytoskeleton | ROCK1, ROCK2 |
| 1615 | CZC24832 | PI3K/mTOR signaling | PI3KG |
| 1632 | Ribociclib | Cell cycle | CDK4, CDK6 |
图8
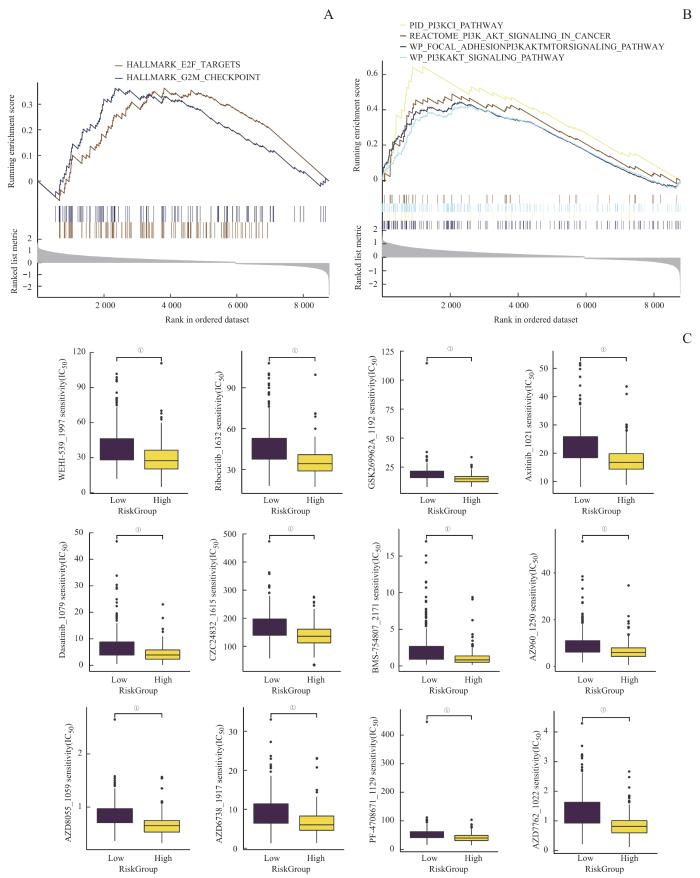
图8
潜在药物预测
Note: A/B. GSEA of potential drug resistance-related pathways. C. A comprehensive analysis of drug sensitivity between the high and low Risk Score groups. ①P<0.001.
Fig 8
Prediction of potential drugs
3 讨论
本研究整合了常规RNA-seq与scRNA-seq数据,以探讨M2巨噬细胞对于PCa预后预测的潜在作用。研究发现,高浸润M2巨噬细胞的PCa患者显著表现出更低的PFS。M2巨噬细胞可以高表达免疫调节、细胞吞噬、细胞趋化相关的基因,与TME中多种免疫细胞相互作用,促进免疫抑制性肿瘤微环境的形成,进而影响肿瘤的发展和预后。因此,本研究基于M2巨噬细胞特征基因构建风险评分模型,模型公式为:RiskScore=0.095 257 59×TREM2+0.081 748 26×OTOA+0.097 203 08×SIGLEC1+0.171 846 15×PLXDC1。该模型在内部测试集与外部验证集中均得到良好验证,说明其预后预测性能具有可重复性和临床适用性。研究表明,高风险评分患者往往有着更差的临床分期与病理分级,同时他们的TME呈现免疫抑制特征,并伴有AR信号通路活性降低,最终表现出更差的预后结果。最后本研究进行药物敏感性分析,筛选出6种对于高风险评分患者疗效更佳的治疗药物。
预后模型中的4个基因在肿瘤研究中日益受到关注。研究[25]表明TREM2+ TAMs的表达水平与肾透明细胞癌的术后复发显著相关。靶向TREM2+ TAMs治疗可以增强TME中T细胞的反应,从而改善对PD-1/PD-L1免疫疗法的治疗抵抗[26-27]。OTOA基因被认为在听力损伤中发挥重要作用,但也可以作为肿瘤的生物标志物[28-30]。SIGLEC1+ TAMs可以与黑色素瘤细胞相互作用,并促进肿瘤早期淋巴结转移[31]。乳腺癌细胞能够上调TAMs上的SIGLEC1表达水平,因此SIGLEC1也被认为是乳腺癌中独立的不良预后指标[32]。而PLXDC1在内皮细胞的血管形态发生中发挥关键作用,被认为与骨肉瘤转移相关,可以作为骨肉瘤的预后指标[33]。这些研究进一步提示了本研究构建的风险评分模型的可靠性,也为之后研究M2巨噬细胞在PCa中的具体作用机制提示了新方向。
尽管本研究取得了重要发现,但仍存在若干局限性。首先,该研究是基于对公共数据库的回顾性分析,尚需进一步的前瞻性临床研究来验证这些发现;其次,尽管构建的预后模型在测试集与验证集中表现良好,但它对于不同患者人群与疾病不同阶段的适用性仍需进一步验证;此外,本研究主要集中于M2巨噬细胞在PCa中的作用,对其他免疫细胞与肿瘤预后之间的关系仍需进行更深入的研究。
综上,本研究通过整合普通RNA-seq与scRNA-seq数据,探索免疫细胞之间的相互作用,并基于多种统计学方法构建了风险评分模型,以预测PCa患者的预后,为PCa个体化治疗与免疫治疗策略提供了重要理论支持。尽管仍需进一步阐明免疫细胞在PCa中的具体作用机制,但该研究为深入理解PCa发病机制及新疗法的开发奠定了基础。
作者贡献声明
韩邦旻负责课题设计与论文修改,汤开然、冯成领负责数据搜集、数据分析、论文撰写与修改,所有作者均阅读并同意了最终稿件的提交。
AUTHOR's CONTRIBUTIONS
HAN Bangmin was responsible for study design and manuscript revision. TANG Kairan and FENG Chengling were responsible for data collection, data analysis, manuscript writing, and revision. All authors have read the last version of paper and consented to its submission.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
All authors disclose no relevant conflict of interests.
参考文献

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}