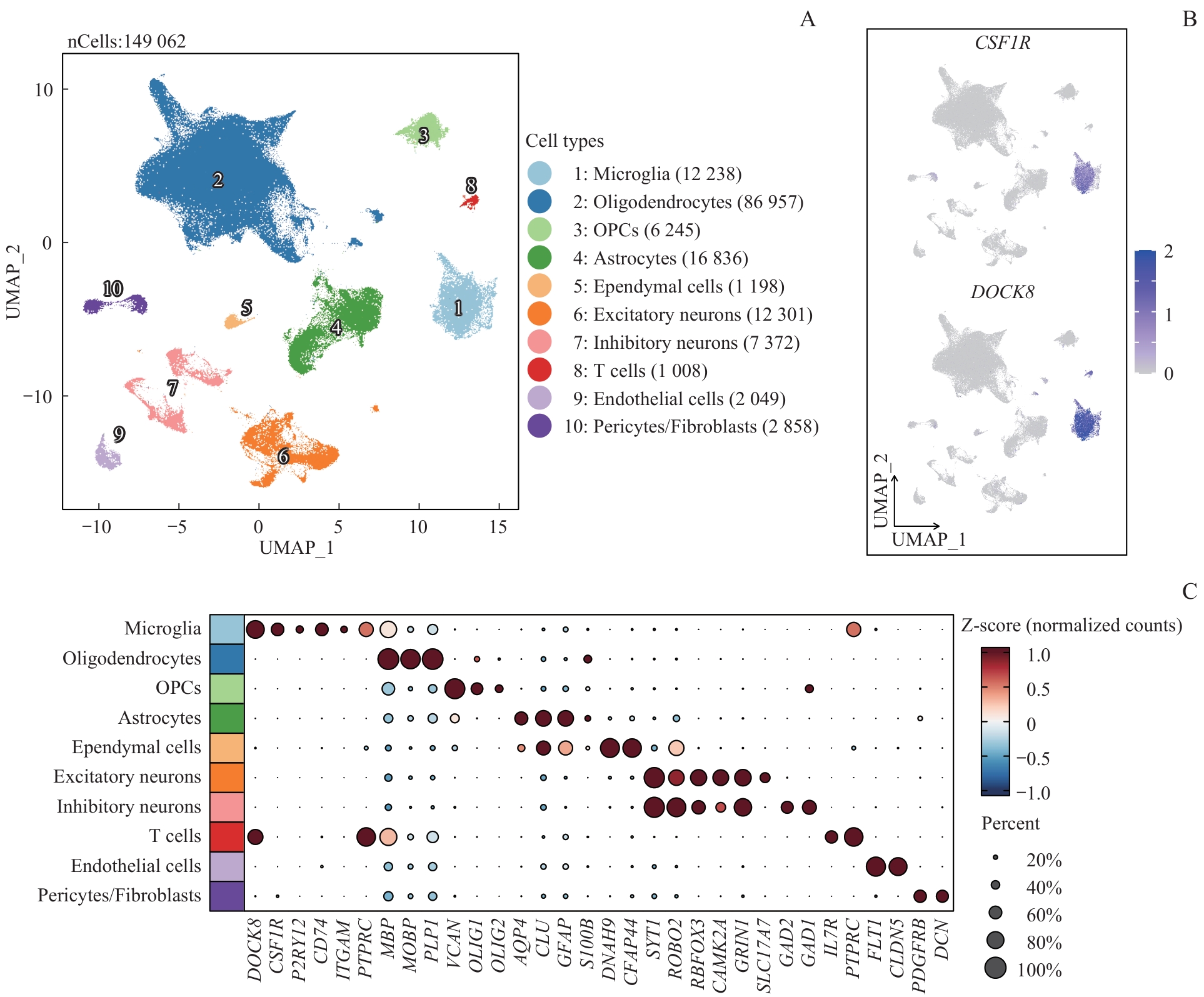
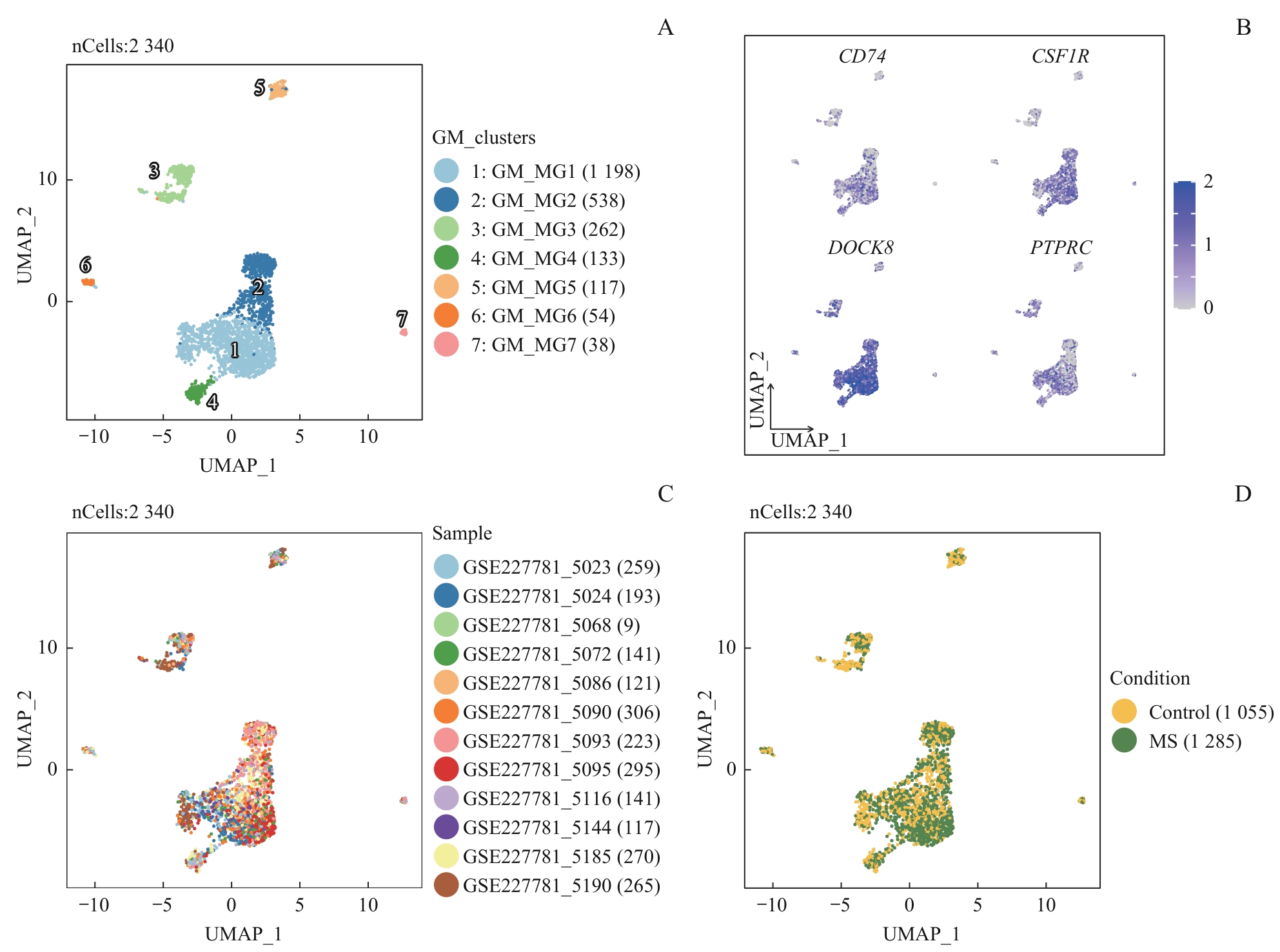
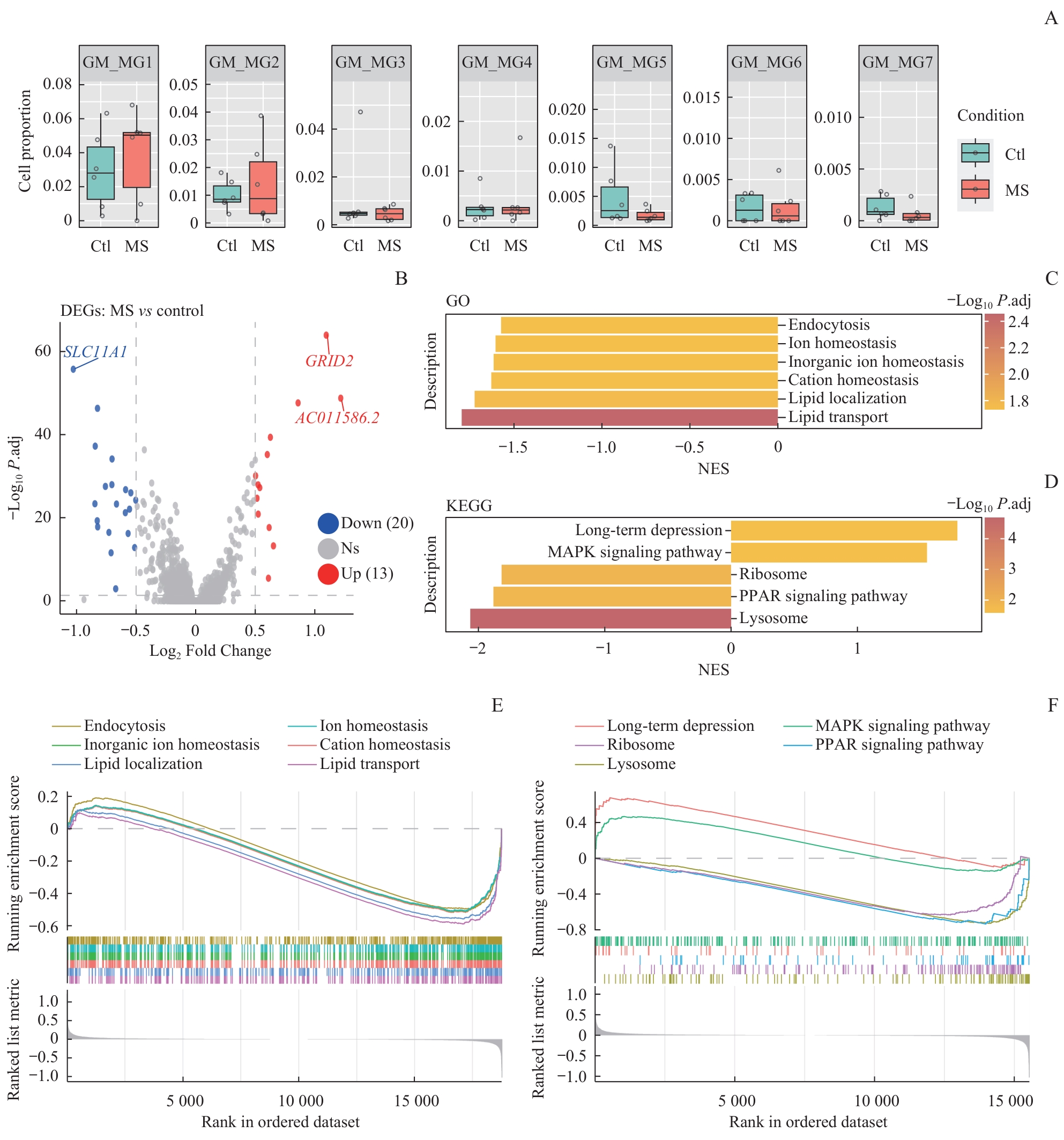
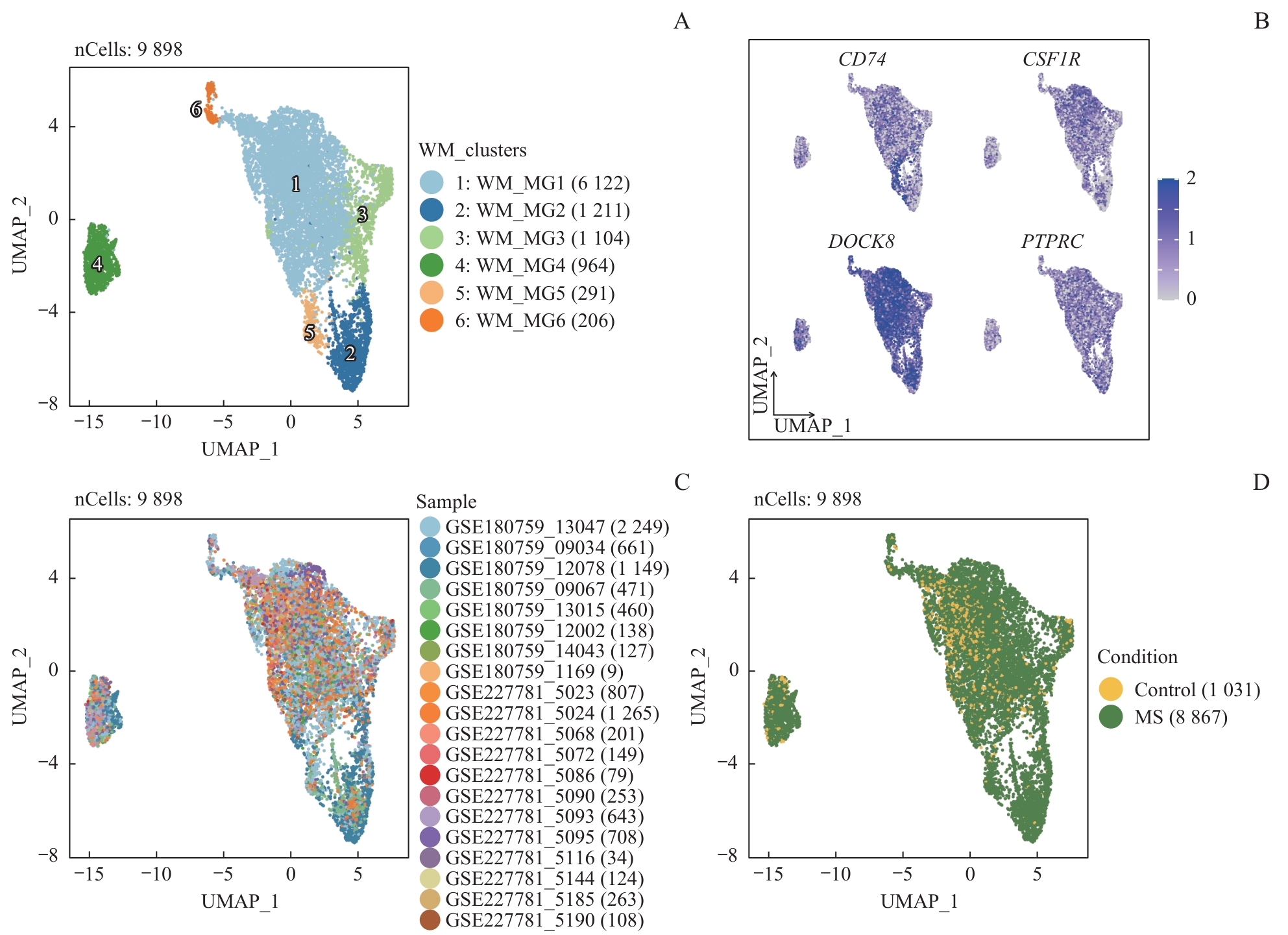
| 1 |
BENEDICT R H B, AMATO M P, DELUCA J, et al. Cognitive impairment in multiple sclerosis: clinical management, MRI, and therapeutic avenues[J]. Lancet Neurol, 2020, 19(10): 860-871.
|
| 2 |
BANWELL B, BENNETT J L, MARIGNIER R, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD Panel proposed criteria[J]. Lancet Neurol, 2023, 22(3): 268-282.
|
| 3 |
KUHLMANN T, MOCCIA M, COETZEE T, et al. Multiple sclerosis progression: time for a new mechanism-driven framework[J]. Lancet Neurol, 2023, 22(1): 78-88.
|
| 4 |
谭红梅, 全超. 多发性硬化疾病修正治疗进展[J]. 重庆医科大学学报, 2024, 49(5): 588-592.
|
|
TAN H M, QUAN C. Advances in disease-modifying therapy for multiple sclerosis[J]. Journal of Chongqing Medical University, 2024, 49(5): 588-592.
|
| 5 |
FAISSNER S, PLEMEL J R, GOLD R, et al. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies[J]. Nat Rev Drug Discov, 2019, 18: 905-922.
|
| 6 |
BORST K, DUMAS A A, PRINZ M. Microglia: immune and non-immune functions[J]. Immunity, 2021, 54(10): 2194-2208.
|
| 7 |
PRINZ M, JUNG S, PRILLER J. Microglia biology: one century of evolving concepts[J]. Cell, 2019, 179(2): 292-311.
|
| 8 |
LUKENS J R, EYO U B. Microglia and neurodevelopmental disorders[J]. Annu Rev Neurosci, 2022, 45: 425-445.
|
| 9 |
DADWAL S, HENEKA M T. Microglia heterogeneity in health and disease[J]. FEBS Open Bio, 2024, 14(2): 217-229.
|
| 10 |
MADORE C, YIN Z, LEIBOWITZ J, et al. Microglia, lifestyle stress, and neurodegeneration[J]. Immunity, 2020, 52(2): 222-240.
|
| 11 |
KENT S A, MIRON V E. Microglia regulation of central nervous system myelin health and regeneration[J]. Nat Rev Immunol, 2024, 24: 49-63.
|
| 12 |
MCNAMARA N B, MUNRO D A D, BESTARD-CUCHE N, et al. Microglia regulate central nervous system myelin growth and integrity[J]. Nature, 2023, 613: 120-129.
|
| 13 |
YONG V W. Microglia in multiple sclerosis: protectors turn destroyers[J]. Neuron, 2022, 110(21): 3534-3548.
|
| 14 |
LLOYD A F, MIRON V E. The pro-remyelination properties of microglia in the central nervous system[J]. Nat Rev Neurol, 2019, 15: 447-458.
|
| 15 |
ZHAO S, UMPIERRE A D, WU L J. Tuning neural circuits and behaviors by microglia in the adult brain[J]. Trends Neurosci, 2024, 47(3): 181-194.
|
| 16 |
SHERAFAT A, PFEIFFER F, REISS A M, et al. Microglial neuropilin-1 promotes oligodendrocyte expansion during development and remyelination by trans-activating platelet-derived growth factor receptor[J]. Nat Commun, 2021, 12: 2265.
|
| 17 |
DONG Y F, D′MELLO C, PINSKY W, et al. Oxidized phosphatidylcholines found in multiple sclerosis lesions mediate neurodegeneration and are neutralized by microglia[J]. Nat Neurosci, 2021, 24: 489-503.
|
| 18 |
LAMPRON A, LAROCHELLE A, LAFLAMME N, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes[J]. J Exp Med, 2015, 212(4): 481-495.
|
| 19 |
RAWJI K S, YOUNG A M H, GHOSH T, et al. Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system[J]. Acta Neuropathol, 2020, 139(5): 893-909.
|
| 20 |
HAGAN N, KANE J L, GROVER D, et al. CSF1R signaling is a regulator of pathogenesis in progressive MS[J]. Cell Death Dis, 2020, 11: 904.
|
| 21 |
HWANG D, SEYEDSADR M S, ISHIKAWA L L W, et al. CSF-1 maintains pathogenic but not homeostatic myeloid cells in the central nervous system during autoimmune neuroinflammation[J]. Proc Natl Acad Sci USA, 2022, 119(14): e2111804119.
|
| 22 |
MARZAN D E, BRÜGGER-VERDON V, WEST B L, et al. Activated microglia drive demyelination via CSF1R signaling[J]. Glia, 2021, 69(6): 1583-1604.
|
| 23 |
TAHMASEBI F, PASBAKHSH P, MORTEZAEE K, et al. Effect of the CSF1R inhibitor PLX3397 on remyelination of corpus callosum in a cuprizone-induced demyelination mouse model[J]. J Cell Biochem, 2019, 120(6): 10576-10586.
|
| 24 |
VOET S, PRINZ M, VAN LOO G. Microglia in central nervous system inflammation and multiple sclerosis pathology[J]. Trends Mol Med, 2019, 25(2): 112-123.
|
| 25 |
SEN M K, MAHNS D A, COORSSEN J R, et al. The roles of microglia and astrocytes in phagocytosis and myelination: insights from the cuprizone model of multiple sclerosis[J]. Glia, 2022, 70(7): 1215-1250.
|
| 26 |
JAKIMOVSKI D, BITTNER S, ZIVADINOV R, et al. Multiple sclerosis[J]. Lancet, 2024, 403(10422):183-202.
|
| 27 |
FRISCHER J M, BRAMOW S, DAL-BIANCO A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains[J]. Brain, 2009, 132(5): 1175-1189.
|
| 28 |
ABSINTA M, MARIC D, GHARAGOZLOO M, et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis[J]. Nature, 2021, 597: 709-714.
|
| 29 |
CLARK I C, GUTIÉRREZ-VÁZQUEZ C, WHEELER M A, et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation[J]. Science, 2021, 372(6540): eabf1230.
|
| 30 |
SCHIRMER L, VELMESHEV D, HOLMQVIST S, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis[J]. Nature, 2019, 573: 75-82.
|
| 31 |
JÄKEL S, AGIRRE E, MENDANHA FALCÃO A, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis[J]. Nature, 2019, 566: 543-547.
|
| 32 |
KISS M G, MINDUR J E, YATES A G, et al. Interleukin-3 coordinates glial-peripheral immune crosstalk to incite multiple sclerosis[J]. Immunity, 2023, 56(7): 1502-1514.e8.
|
| 33 |
MIEDEMA A, GERRITS E, BROUWER N, et al. Brain macrophages acquire distinct transcriptomes in multiple sclerosis lesions and normal appearing white matter[J]. Acta Neuropathol Commun, 2022, 10(1): 8.
|
| 34 |
HAO Y, HAO S, ANDERSEN-NISSEN E, et al. Integrated analysis of multimodal single-cell data[J]. Cell, 2021, 184(13): 3573-3587.e29.
|
| 35 |
HAFEMEISTER C, SATIJA R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression[J]. Genome Biol, 2019, 20(1): 296.
|
| 36 |
KORSUNSKY I, MILLARD N, FAN J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony[J]. Nat Meth, 2019, 16: 1289-1296.
|
| 37 |
HU C, LI T, XU Y, et al. CellMarker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data[J]. Nucleic Acids Res, 2023, 51(d1): D870-D876.
|
| 38 |
WU T, HU E, XU S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data[J]. Innovation (Camb), 2021, 2(3): 100141.
|
| 39 |
YU G, WANG L G, HAN Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters[J]. OMICS, 2012, 16(5): 284-287.
|
| 40 |
TRAPNELL C, CACCHIARELLI D, GRIMSBY J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells[J]. Nat Biotechnol, 2014, 32: 381-386.
|
| 41 |
QIU X J, HILL A, PACKER J, et al. Single-cell mRNA quantification and differential analysis with Census[J]. Nat Meth, 2017, 14: 309-315.
|
| 42 |
QIU X J, MAO Q, TANG Y, et al. Reversed graph embedding resolves complex single-cell trajectories[J]. Nat Meth, 2017, 14: 979-982.
|
| 43 |
CAO J Y, SPIELMANN M, QIU X J, et al. The single-cell transcriptional landscape of mammalian organogenesis[J]. Nature, 2019, 566: 496-502.
|
| 44 |
AIBAR S, GONZÁLEZ-BLAS C B, MOERMAN T, et al. SCENIC: single-cell regulatory network inference and clustering[J]. Nat Meth, 2017, 14: 1083-1086.
|
| 45 |
HÄNZELMANN S, CASTELO R, GUINNEY J. GSVA: gene set variation analysis for microarray and RNA-seq data[J]. BMC Bioinformatics, 2013, 14: 7.
|
| 46 |
RITCHIE M E, PHIPSON B, WU D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies[J]. Nucleic Acids Res, 2015, 43(7): e47.
|
| 47 |
KOOI E J, STRIJBIS E M, VAN DER VALK P, et al. Heterogeneity of cortical lesions in multiple sclerosis: clinical and pathologic implications[J]. Neurology, 2012, 79(13): 1369-1376.
|
| 48 |
SINGHAL T, O′CONNOR K, DUBEY S, et al. Gray matter microglial activation in relapsing vs progressive MS: a[F-18]PBR06-PET study[J]. Neurol Neuroimmunol Neuroinflamm, 2019, 6(5): e587.
|
| 49 |
FAN L Y, YANG J, LIU R Y, et al. Integrating single-nucleus sequence profiling to reveal the transcriptional dynamics of Alzheimer′s disease, Parkinson's disease, and multiple sclerosis[J]. J Transl Med, 2023, 21(1): 649.
|
| 50 |
ZINATIZADEH M R, SCHOCK B, CHALBATANI G M, et al. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases[J]. Genes Dis, 2021, 8(3): 287-297.
|
 ), 孙锋1,2,3, 吴文玉1,2,3, 邵付明4, 高正良1,2,3,4(
), 孙锋1,2,3, 吴文玉1,2,3, 邵付明4, 高正良1,2,3,4(