Journal of Shanghai Jiao Tong University (Medical Science) ›› 2025, Vol. 45 ›› Issue (10): 1308-1319.doi: 10.3969/j.issn.1674-8115.2025.10.006
• Basic research • Previous Articles Next Articles
YU Zhiyuan1, DONG Haiping1,2, GAO Nan1, MA Ke1( )
)
Received:2025-05-28
Accepted:2025-07-17
Online:2025-10-28
Published:2025-10-28
Contact:
MA Ke
E-mail:marke72@163.com
Supported by:CLC Number:
YU Zhiyuan, DONG Haiping, GAO Nan, MA Ke. Identification and mechanistic analysis of core genes associated with morphine tolerance in dorsal root ganglion: an integrative transcriptomics approach using WGCNA and machine learning algorithms[J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(10): 1308-1319.
Add to citation manager EndNote|Ris|BibTeX
URL: https://xuebao.shsmu.edu.cn/EN/10.3969/j.issn.1674-8115.2025.10.006
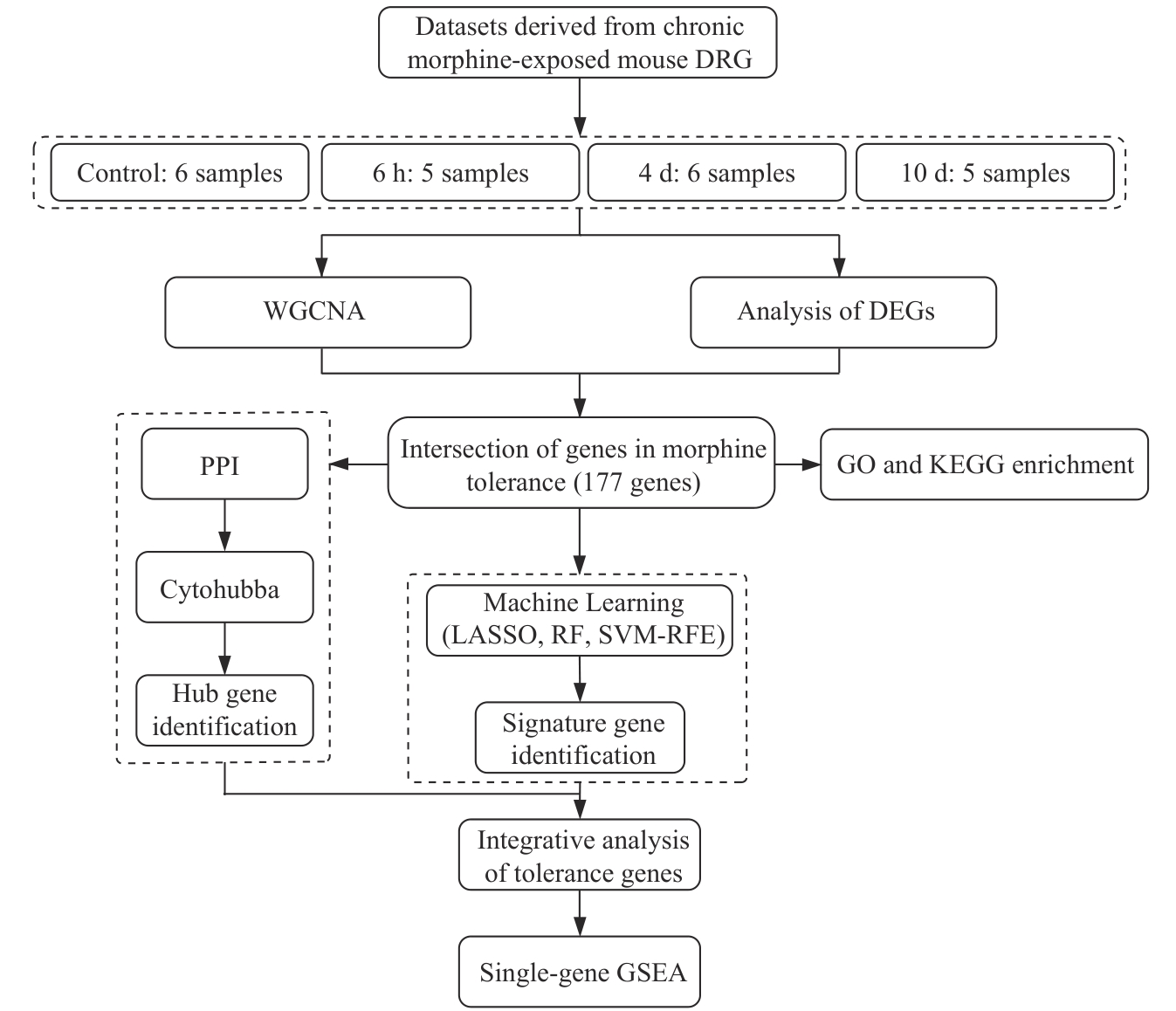
Fig 1 Bioinformatics analysis workflow
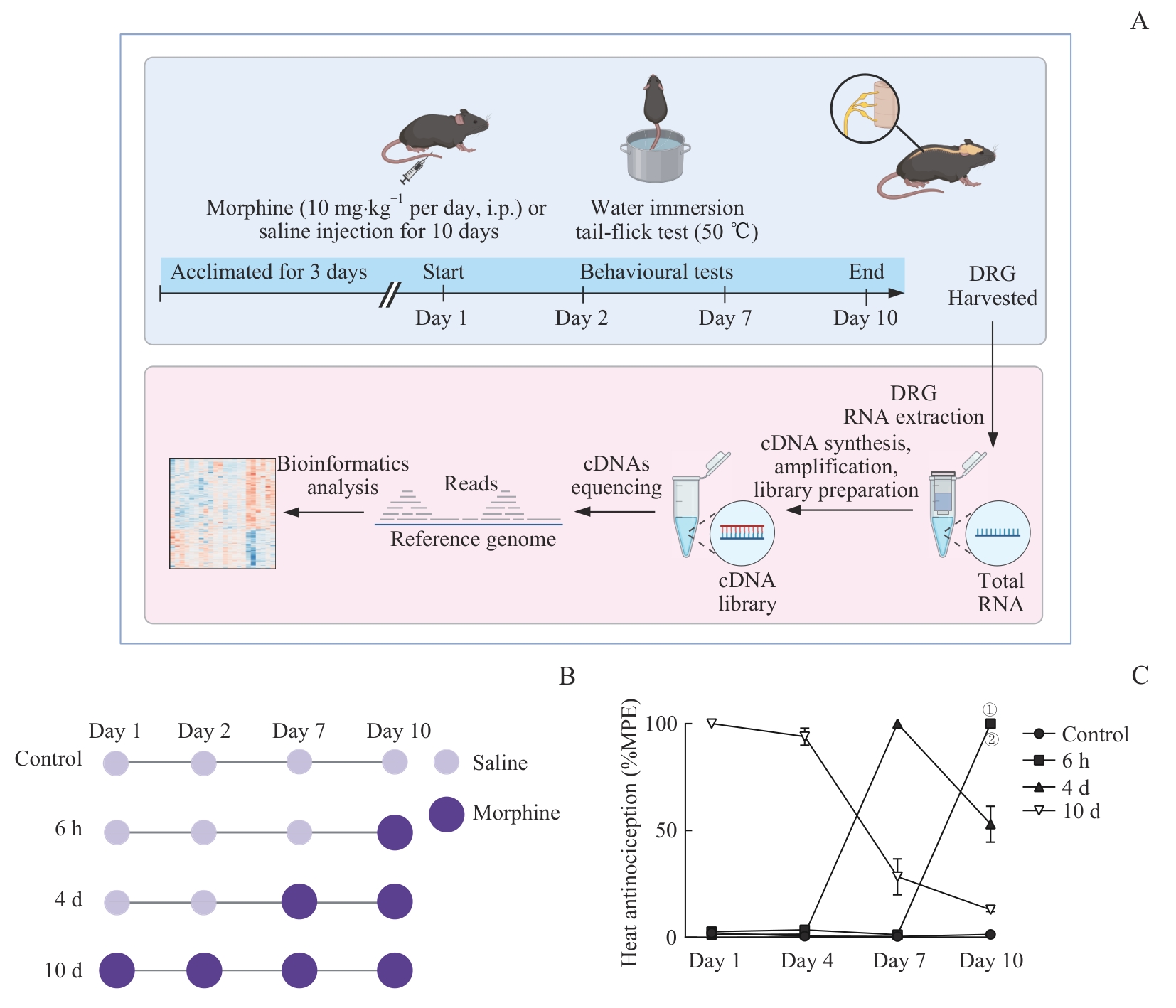
Fig 2 Establishment and behavioral evaluation of mouse models with different morphine administration durations
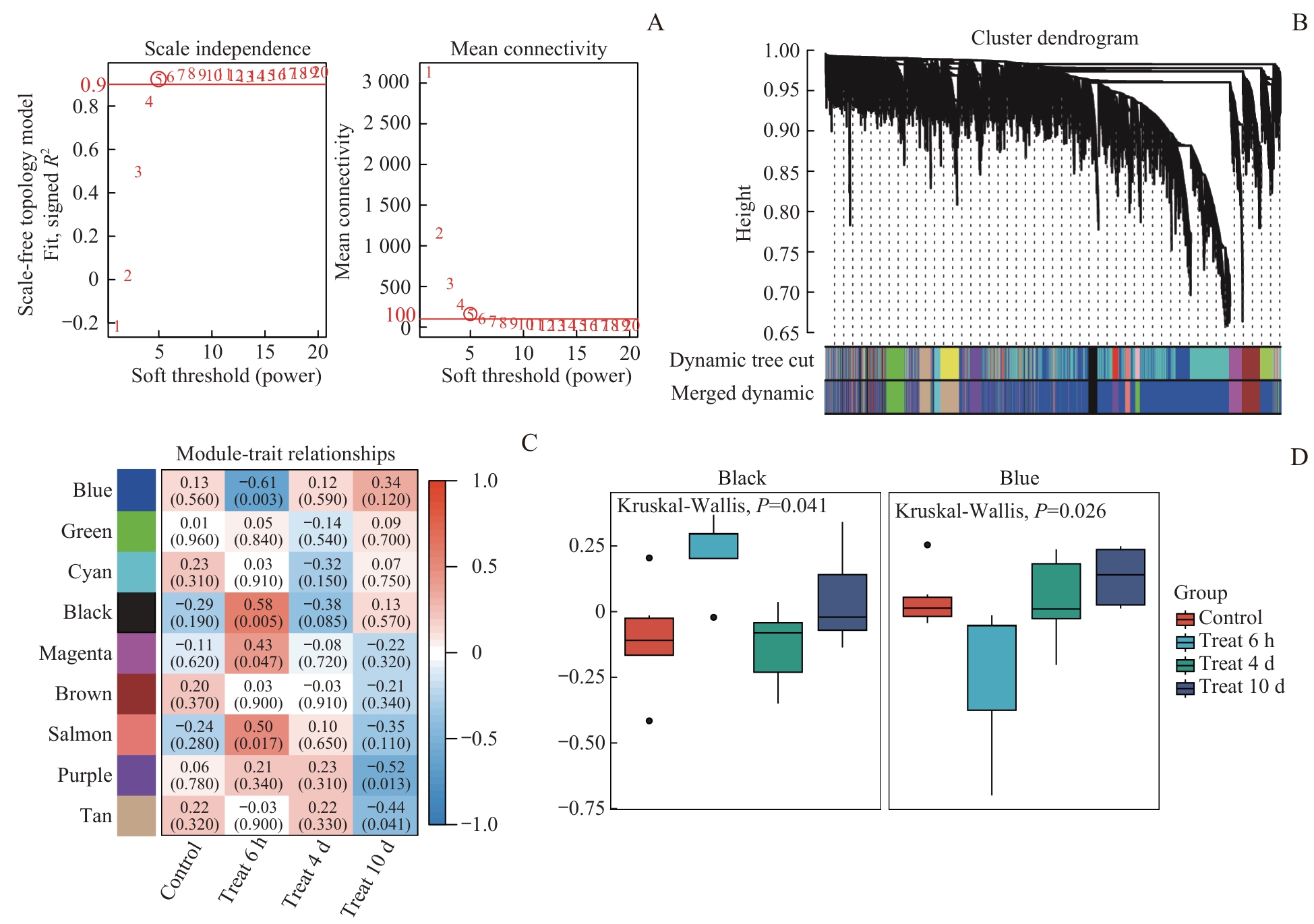
Fig 3 Identification of key morphine tolerance-associated gene modules by WGCNA
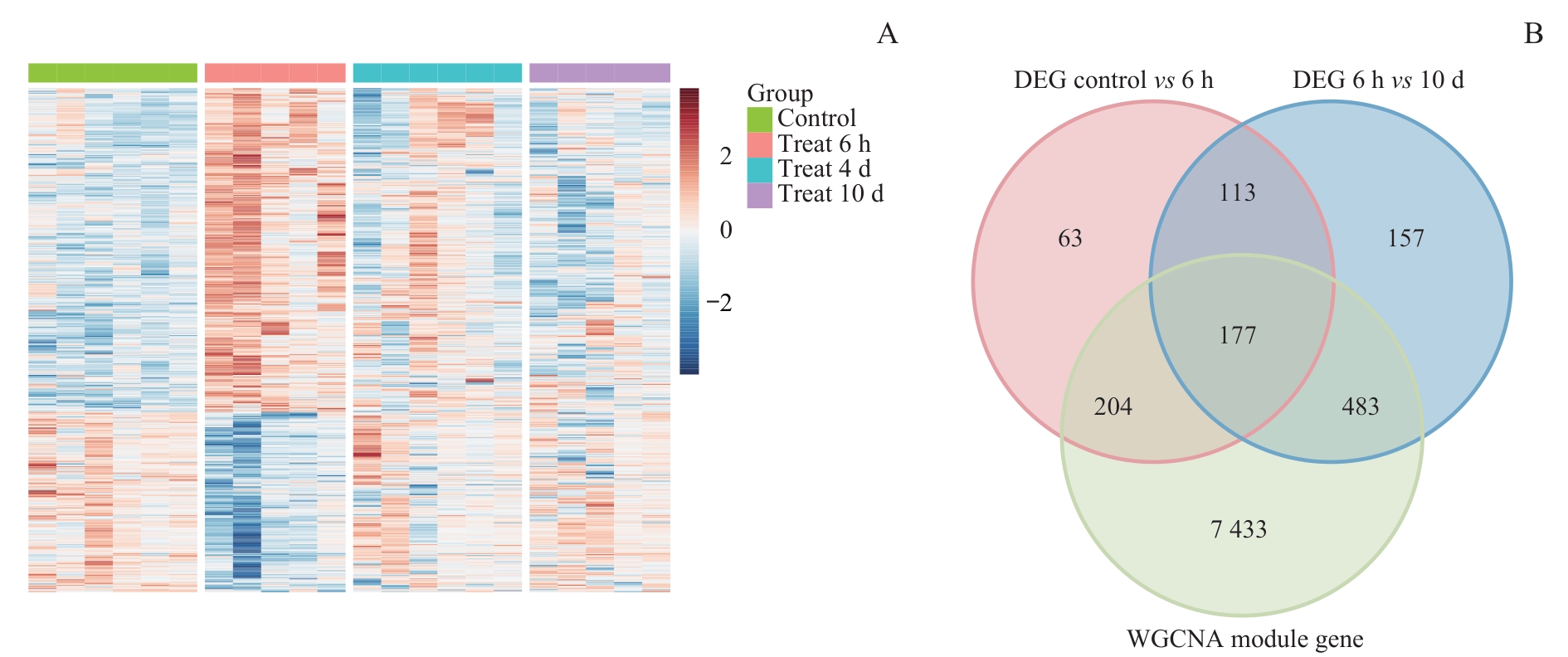
Fig 4 Intersection analysis of morphine tolerance-associated DEGs and WGCNA module genes
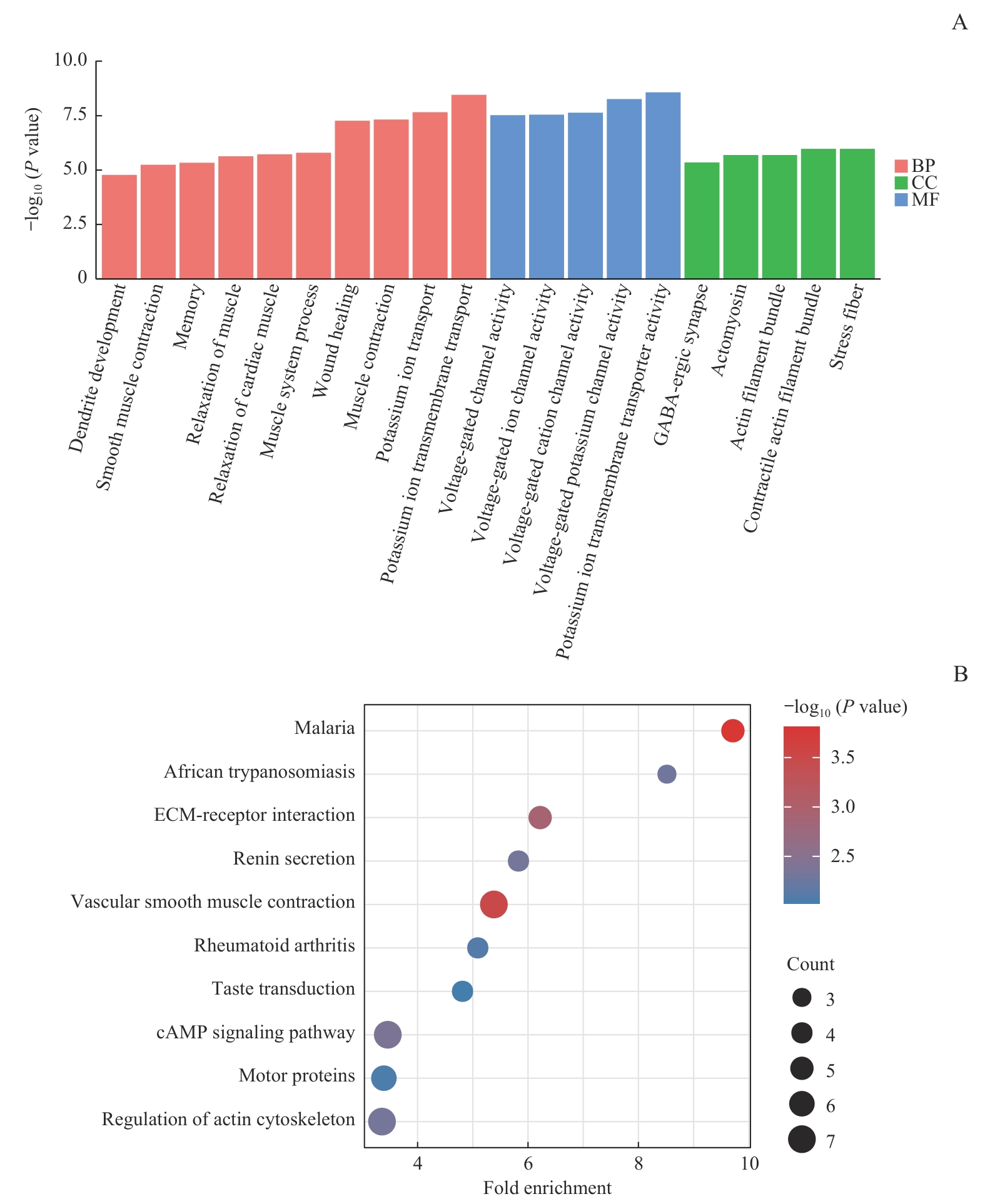
Fig 5 GO and KEGG enrichment analysis of candidate genes
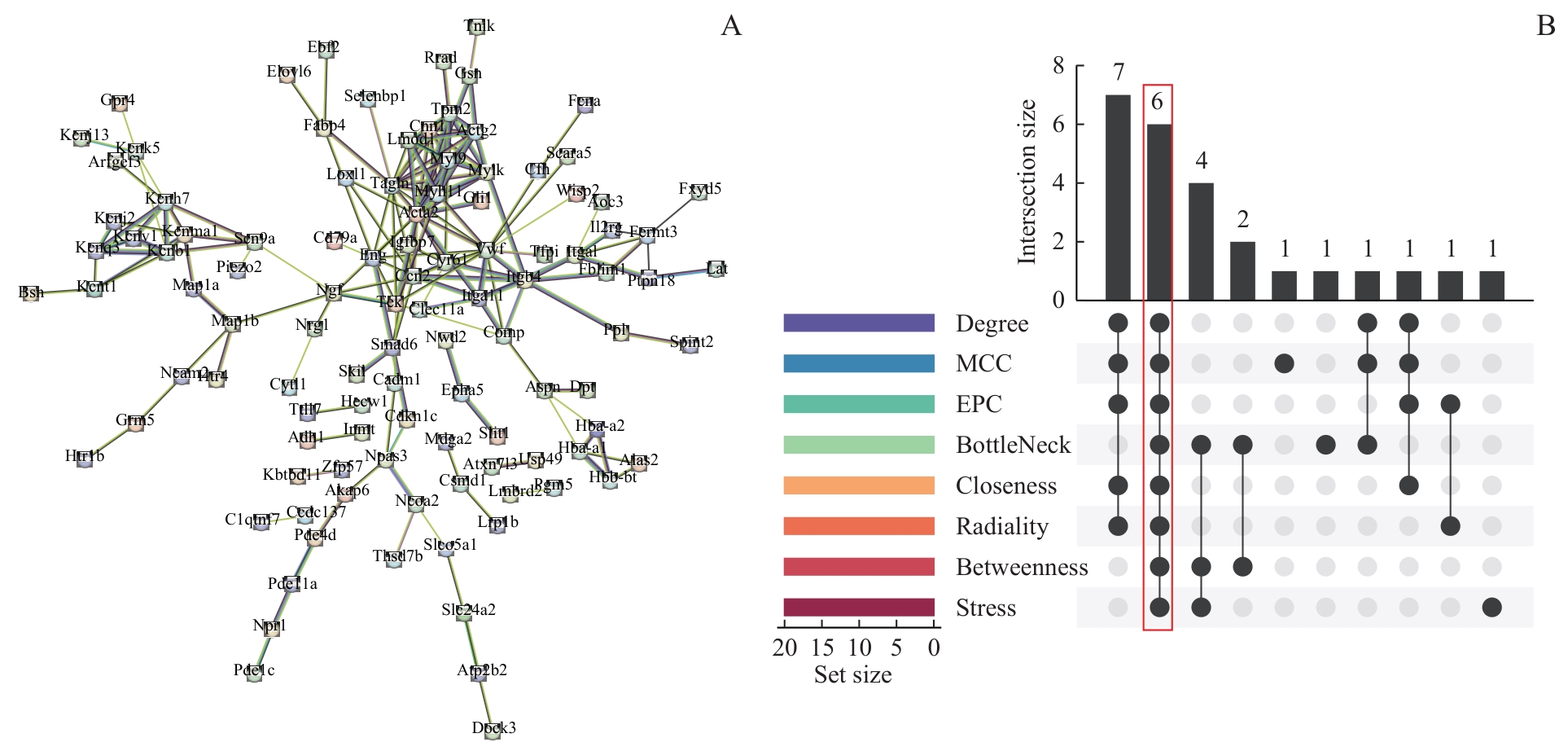
Fig 6 PPI network and screening of hub genes
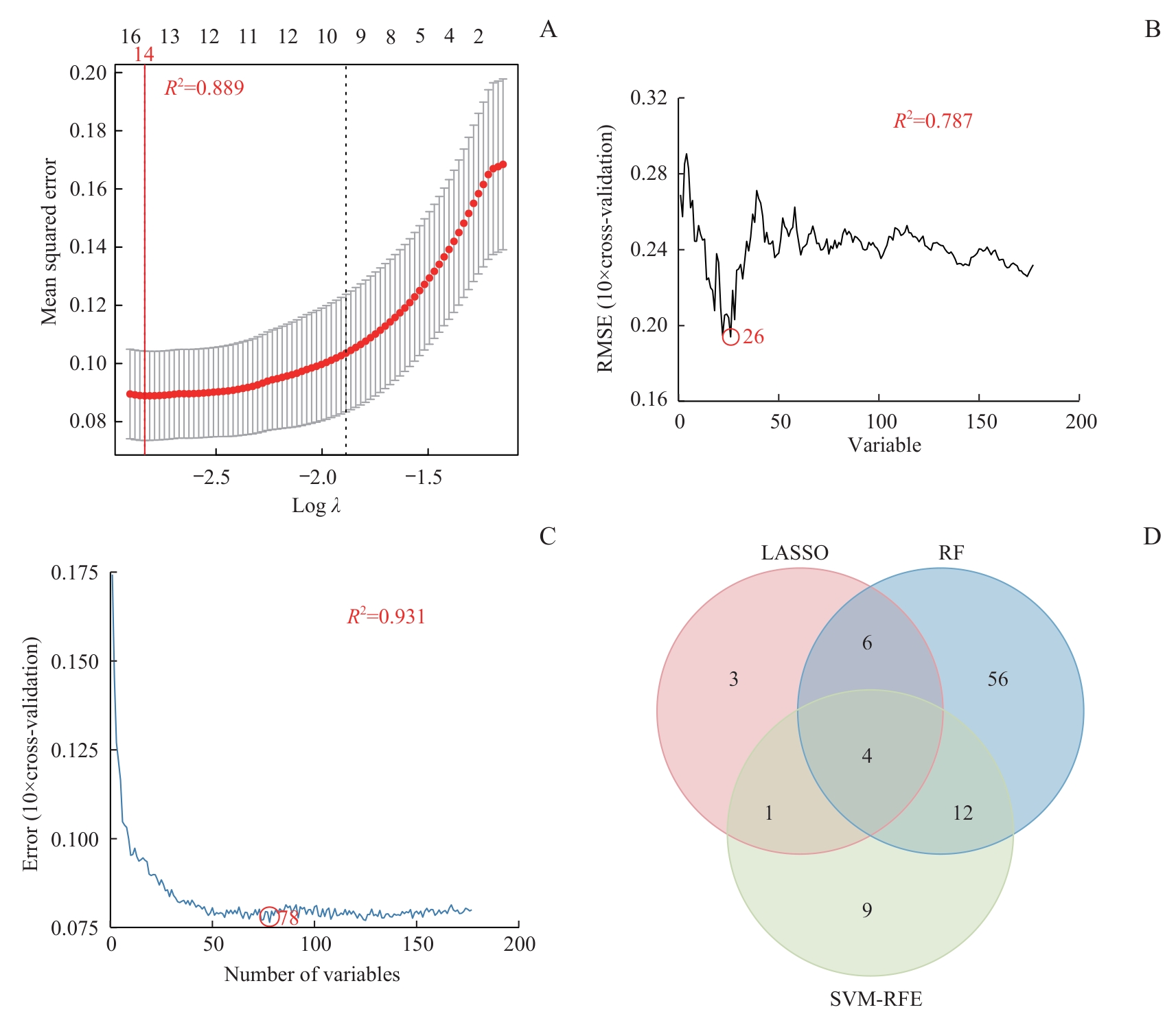
Fig 7 Screening and integration of signature genes
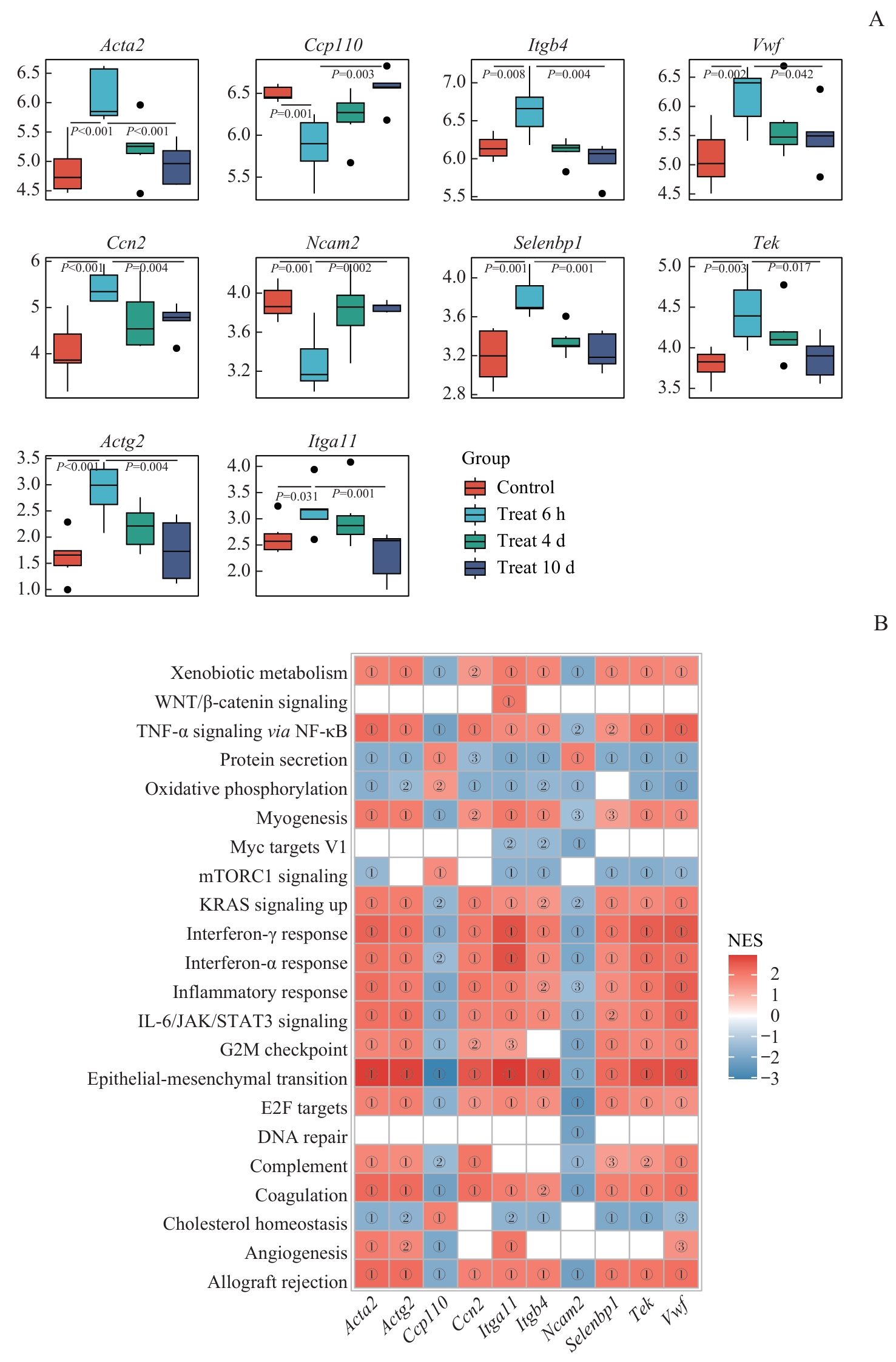
Fig 8 Intergroup expression differences of hub and signature genes and their GSEA enrichment associations
| [1] | RUEDA-RUZAFA L, CRUZ F, CARDONA D, et al. Opioid system influences gut-brain axis: dysbiosis and related alterations[J]. Pharmacol Res, 2020, 159: 104928. |
| [2] | TAYLOR J L, SAMET J H. Opioid use disorder[J]. Ann Intern Med, 2022, 175(1): ITC1-ITC16. |
| [3] | CHOE K, ZINN E, LU K, et al. Impact of COVID-19 pandemic on chronic pain and opioid use in marginalized populations: a scoping review[J]. Front Public Health, 2023, 11: 1046683. |
| [4] | LEMOS DUARTE M, DEVI L A. Post-translational modifications of opioid receptors[J]. Trends Neurosci, 2020, 43(6): 417-432. |
| [5] | LI L, CHEN J, LI Y Q. The downregulation of opioid receptors and neuropathic pain[J]. Int J Mol Sci, 2023, 24(6): 5981. |
| [6] | OCHANDARENA N E, NIEHAUS J K, TASSOU A, et al. Cell-type specific molecular architecture for mu opioid receptor function in pain and addiction circuits[J]. Neuropharmacology, 2023, 238: 109597. |
| [7] | DUNN A D, ROBINSON S A, NWOKAFOR C, et al. Molecular and long-term behavioral consequences of neonatal opioid exposure and withdrawal in mice[J]. Front Behav Neurosci, 2023, 17: 1202099. |
| [8] | RUIVO J, TAVARES I, POZZA D H. Molecular targets in bone cancer pain: a systematic review of inflammatory cytokines[J]. J Mol Med (Berl), 2024, 102(9): 1063-1088. |
| [9] | BI K, LEI Y, KONG D, et al. Progress in the study of intestinal microbiota involved in morphine tolerance[J]. Heliyon, 2024, 10(6): e27187. |
| [10] | BERTA T, STRONG J A, ZHANG J M, et al. Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain: an update[J]. Expert Opin Ther Targets, 2023, 27(8): 665-678. |
| [11] | QUIRION B, BEAULIEU C, CÔTÉ L, et al. Distribution of delta and mu opioid receptor mRNA in rodent dorsal root ganglia neurons[J]. Eur J Neurosci, 2022, 56(3): 4031-4044. |
| [12] | RUIZ-CANTERO M C, CORTÉS-MONTERO E, JAIN A, et al. The sigma-1 receptor curtails endogenous opioid analgesia during sensitization of TRPV1 nociceptors[J]. Br J Pharmacol, 2023, 180(8): 1148-1167. |
| [13] | FÜRST S, ZÁDORI Z S, ZÁDOR F, et al. On the role of peripheral sensory and gut mu opioid receptors: peripheral analgesia and tolerance[J]. Molecules, 2020, 25(11): 2473. |
| [14] | WANG B, JIANG B W, LI G W, et al. Somatosensory neurons express specific sets of lincRNAs, and lincRNA CLAP promotes itch sensation in mice[J]. EMBO Rep, 2023, 24(2): e54313. |
| [15] | WANG K K, WANG S S, CHEN Y, et al. Single-cell transcriptomic analysis of somatosensory neurons uncovers temporal development of neuropathic pain[J]. Cell Res, 2021, 31(8): 904-918. |
| [16] | KUPARI J, USOSKIN D, PARISIEN M, et al. Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain[J]. Nat Commun, 2021, 12(1): 1510. |
| [17] | KONG X J, SUN H R, WEI K M, et al. WGCNA combined with machine learning algorithms for analyzing key genes and immune cell infiltration in heart failure due to ischemic cardiomyopathy[J]. Front Cardiovasc Med, 2023, 10: 1058834. |
| [18] | CHEN Y M, LIU F, SHI S N, et al. The integrated transcriptome bioinformatics analysis of energy metabolism-related profiles for dorsal root ganglion of neuropathic pain[J]. Mol Neurobiol, 2025, 62(4): 4149-4171. |
| [19] | RIVAT C, SEBAIHI S, VAN STEENWINCKEL J, et al. Src family kinases involved in CXCL12-induced loss of acute morphine analgesia[J]. Brain Behav Immun, 2014, 38: 38-52. |
| [20] | STRANG J, VOLKOW N D, DEGENHARDT L, et al. Opioid use disorder[J]. Nat Rev Dis Primers, 2020, 6(1): 3. |
| [21] | SAKLOTH F, POLIZU C, BERTHERAT F, et al. Regulators of G protein signaling in analgesia and addiction[J]. Mol Pharmacol, 2020, 98(6): 739-750. |
| [22] | MASUHO I, BALAJI S, MUNTEAN B S, et al. A global map of G protein signaling regulation by RGS proteins[J]. Cell, 2020, 183(2): 503-521.e19. |
| [23] | HOU X R, WENG Y Q, GUO Q L, et al. Transcriptomic analysis of long noncoding RNAs and mRNAs expression profiles in the spinal cord of bone cancer pain rats[J]. Mol Brain, 2020, 13(1): 47. |
| [24] | FALCONNIER C, CAPARROS-ROISSARD A, DECRAENE C, et al. Functional genomic mechanisms of opioid action and opioid use disorder: a systematic review of animal models and human studies[J]. Mol Psychiatry, 2023, 28(11): 4568-4584. |
| [25] | COLVIN L A, BULL F, HALES T G. Perioperative opioid analgesia-when is enough too much? A review of opioid-induced tolerance and hyperalgesia[J]. Lancet, 2019, 393(10180): 1558-1568. |
| [26] | GAMBLE M C, WILLIAMS B R, SINGH N, et al. Mu-opioid receptor and receptor tyrosine kinase crosstalk: implications in mechanisms of opioid tolerance, reduced analgesia to neuropathic pain, dependence, and reward[J]. Front Syst Neurosci, 2022, 16: 1059089. |
| [27] | MARTUCCI K T. Neuroimaging of opioid effects in humans across conditions of acute administration, chronic pain therapy, and opioid use disorder[J]. Trends Neurosci, 2024, 47(6): 418-431. |
| [28] | GARCÍA-DOMÍNGUEZ M. Enkephalins and pain modulation: mechanisms of action and therapeutic perspectives[J]. Biomolecules, 2024, 14(8): 926. |
| [29] | NGUYEN D, NGUYEN H, ONG H, et al. Ensemble learning using traditional machine learning and deep neural network for diagnosis of Alzheimer's disease[J]. IBRO Neurosci Rep, 2022, 13: 255-263. |
| [30] | DE ANGELI K, GAO S, BLANCHARD A, et al. Using ensembles and distillation to optimize the deployment of deep learning models for the classification of electronic cancer pathology reports[J]. JAMIA Open, 2022, 5(3): ooac075. |
| [31] | TAKEFUJI Y. Beyond XGBoost and SHAP: unveiling true feature importance[J]. J Hazard Mater, 2025, 488: 137382. |
| [32] | ZHANG W Y, CHEN Z H, AN X X, et al. Analysis and validation of diagnostic biomarkers and immune cell infiltration characteristics in pediatric sepsis by integrating bioinformatics and machine learning[J]. World J Pediatr, 2023, 19(11): 1094-1103. |
| [33] | COBOS E J, NICKERSON C A, GAO F Y, et al. Mechanistic differences in neuropathic pain modalities revealed by correlating behavior with global expression profiling[J]. Cell Rep, 2018, 22(5): 1301-1312. |
| [34] | XIE S W, NASLAVSKY N, CAPLAN S. Emerging insights into CP110 removal during early steps of ciliogenesis[J]. J Cell Sci, 2024, 137(4): jcs261579. |
| [35] | SONG T, YANG Y F, ZHOU P, et al. ENKD1 promotes CP110 removal through competing with CEP97 to initiate ciliogenesis[J]. EMBO Rep, 2022, 23(5): e54090. |
| [36] | NISHIMURA Y, KASAHARA K, SHIROMIZU T, et al. Primary cilia as signaling hubs in health and disease[J]. Adv Sci (Weinh), 2018, 6(1): 1801138. |
| [37] | WACHTEN D, MICK D U. Signal transduction in primary cilia: analyzing and manipulating GPCR and second messenger signaling[J]. Pharmacol Ther, 2021, 224: 107836. |
| [38] | HILL S A, FU M, GARCIA A D R. Sonic hedgehog signaling in astrocytes[J]. Cell Mol Life Sci, 2021, 78(4): 1393-1403. |
| [39] | MA R, KUTCHY N A, WANG Z B, et al. Extracellular vesicle-mediated delivery of anti-miR-106b inhibits morphine-induced primary ciliogenesis in the brain[J]. Mol Ther, 2023, 31(5): 1332-1345. |
| [40] | MA R, KUTCHY N A, HU G K. Astrocyte-derived extracellular vesicle-mediated activation of primary ciliary signaling contributes to the development of morphine tolerance[J]. Biol Psychiatry, 2021, 90(8): 575-585. |
| [41] | MELROSE J, HAYES A J, BIX G. The CNS/PNS extracellular matrix provides instructive guidance cues to neural cells and neuroregulatory proteins in neural development and repair[J]. Int J Mol Sci, 2021, 22(11): 5583. |
| [42] | IBÁÑEZ C F, PARATCHA G, LEDDA F. RET-independent signaling by GDNF ligands and GFRα receptors[J]. Cell Tissue Res, 2020, 382(1): 71-82. |
| [43] | PARCERISAS A, ORTEGA-GASCÓ A, PUJADAS L, et al. The hidden side of NCAM family: NCAM2, a key cytoskeleton organization molecule regulating multiple neural functions[J]. Int J Mol Sci, 2021, 22(18): 10021. |
| [44] | RAWAL P, ZHAO L Q. Sialometabolism in brain health and Alzheimer's disease[J]. Front Neurosci, 2021, 15: 648617. |
| [45] | SUZUKI M, NARITA M, NARITA M, et al. Chronic morphine treatment increases the expression of the neural cell adhesion molecule in the dorsal horn of the mouse spinal cord[J]. Neurosci Lett, 2006, 399(3): 202-205. |
| [46] | EL MAAROUF A, KOLESNIKOV Y, PASTERNAK G, et al. Removal of polysialylated neural cell adhesion molecule increases morphine analgesia and interferes with tolerance in mice[J]. Brain Res, 2011, 1404: 55-62. |
| [47] | RYMUT H E, RUND L A, SOUTHEY B R, et al. Prefrontal cortex response to prenatal insult and postnatal opioid exposure[J]. Genes (Basel), 2022, 13(8): 1371. |
| [48] | SLACK R J, MACDONALD S J F, ROPER J A, et al. Emerging therapeutic opportunities for integrin inhibitors[J]. Nat Rev Drug Discov, 2022, 21(1): 60-78. |
| [49] | CIECHANOWSKA A, ROJEWSKA E, PIOTROWSKA A, et al. New insights into the analgesic properties of the XCL1/XCR1 and XCL1/ITGA9 axes modulation under neuropathic pain conditions-evidence from animal studies[J]. Front Immunol, 2022, 13: 1058204. |
| [50] | PARK J, LEE C, KIM Y T. Effects of natural product-derived compounds on inflammatory pain via regulation of microglial activation[J]. Pharmaceuticals (Basel), 2023, 16(7): 941. |
| [51] | LI H Y, WATKINS L R, WANG X H. Microglia in neuroimmunopharmacology and drug addiction[J]. Mol Psychiatry, 2024, 29(6): 1912-1924. |
| [52] | PAHAN P, XIE J Y. Microglial inflammation modulates opioid analgesic tolerance[J]. J Neurosci Res, 2023, 101(9): 1383-1392. |
| [53] | JALODIA R, ABU Y F, OPPENHEIMER M R, et al. Opioid use, gut dysbiosis, inflammation, and the nervous system[J]. J Neuroimmune Pharmacol, 2022, 17(1/2): 76-93. |
| [1] | Zhu Menglin, Liu Xiao, Xu Xiaodan, Wang Ganhong, Xia Kaijian, Chen Jian. Prediction of delayed post-polypectomy bleeding using a multimodal model [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2026, 46(4): 509-520. |
| [2] | Wang Junxiao, Wang Yuze, Zhang Shasha, Yuan Yu, Zhang Bing, Li Ruogu. Analysis of cardiac features in Lmna N195K mutant mice using single-nucleus RNA sequencing [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2026, 46(3): 275-290. |
| [3] | HUANG Xin, LIU Jiahui, YE Jingwen, QIAN Wenli, XU Wanxing, WANG Lin. Development and clinical application of a machine learning-driven model for metabolite-based diagnosis of small cell lung cancer [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(8): 1009-1016. |
| [4] | HUANG Zihan, HUANG Xinzhi. Application of single-cell RNA sequencing in bone regeneration [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(8): 1053-1058. |
| [5] | ZHU Hanyi, SHI Huan, YU Chuangqi, ZHENG Lingyan. Preliminary study on early warning value and mechanism of interleukin-1β in extremely severe oral and maxillofacial space infections [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(6): 661-672. |
| [6] | ZHANG Xingyu, LI Ruogu. Systematic analysis and exploration of single-cell transcriptomes in aortic aneurysm [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(6): 735-744. |
| [7] | LI Linying, CAI Xiaodong, TONG Ran, YANG Chen, WANG Zhiming, HE Xiaoyu, MA Ziyue, ZHANG Feng, LI Lingjie, ZHOU Junmei. Analysis of transcriptome and chromatin accessibility changes during the differentiation of human embryonic stem cells into neural progenitor cells [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(4): 387-403. |
| [8] | RUAN Qingqing, SU Shuzhi, LI Yanting, REN Yuan, DAI Yong, QIAO Zengyong. Intraoperative complications in percutaneous coronary intervention for acute myocardial infarction: development of a risk prediction model [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(12): 1589-1597. |
| [9] | CAI Qiangwei, SUN Feng, WU Wenyu, SHAO Fuming, GAO Zhengliang, JIN Shengkai. Transcriptional regulatory network analysis of microglia in multiple sclerosis [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(1): 29-41. |
| [10] | WU Qizhen, LIU Qiming, CHAI Yezi, TAO Zhengyu, WANG Yinan, GUO Xinning, JIANG Meng, PU Jun. Evaluation of machine learning prediction of altered inflammatory metabolic state after neoadjuvant therapy for breast cancer [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(9): 1169-1181. |
| [11] | XU Wanxing, WANG Lin, GUO Qiaomei, WANG Xueqing, LOU Jiatao. Clinical validation and application value exploration of multi-modal pulmonary nodule diagnosis model [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(8): 1030-1036. |
| [12] | XU Wenhui, YANG Chang, LI Ruiqing, BIAN Jing, LI Xiayi, ZHENG Leizhen. Exploratory study of interferon regulatory factor 3 promoting proliferation and invasion related to colorectal cancer cells [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(3): 301-311. |
| [13] | SI Chunying, WANG Jianru, LI Xiaohui, WANG Yongxia, GUAN Huaimin. Study on intercellular communication and key genes of smooth muscle cells in human coronary atherosclerosis based on single cell sequencing technology [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(2): 169-182. |
| [14] | LIU Siyu, ZHANG Lei. Sevoflurane inhibits the differentiation and development of neural progenitor cells into neurons in the prefrontal cortex of newborn mice [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2023, 43(9): 1115-1130. |
| [15] | MA Ben, ZHAO Cheng, SHU Yijun, DONG Ping. Application progress of CT radiomics in gastrointestinal stromal tumor [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2023, 43(7): 923-930. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||