JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE) ›› 2021, Vol. 41 ›› Issue (5): 571-578.doi: 10.3969/j.issn.1674-8115.2021.05.003
• Basic research • Previous Articles Next Articles
Lu-di YANG1( ), Gao-ming WANG1, Ren-hao HU2, Xiao-hua JIANG2, Ran CUI2()
), Gao-ming WANG1, Ren-hao HU2, Xiao-hua JIANG2, Ran CUI2()
Online:2021-05-28
Published:2021-05-27
Contact:
Ran CUI
E-mail:yangludi@sjtu.edu.cn;cuiangus@tongji.edu.cn
Supported by:CLC Number:
Lu-di YANG, Gao-ming WANG, Ren-hao HU, Xiao-hua JIANG, Ran CUI. Identification of core genes in pancreatic cancer progression by bioinformatics analysis[J]. JOURNAL OF SHANGHAI JIAOTONG UNIVERSITY (MEDICAL SCIENCE), 2021, 41(5): 571-578.
Add to citation manager EndNote|Ris|BibTeX
URL: https://xuebao.shsmu.edu.cn/EN/10.3969/j.issn.1674-8115.2021.05.003
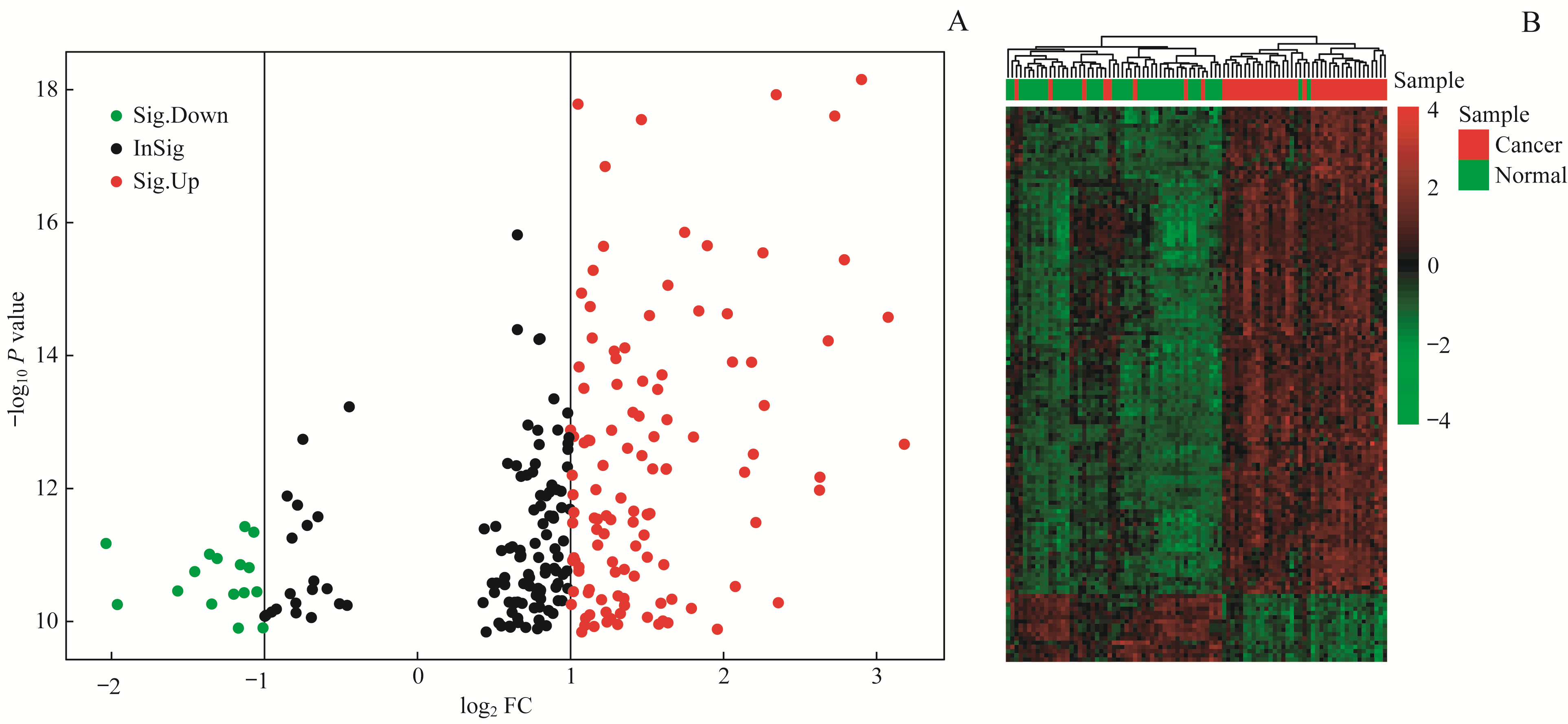
Fig 1 Visual analysis of the DEGs in pancreatic cancer and adjacent tissues
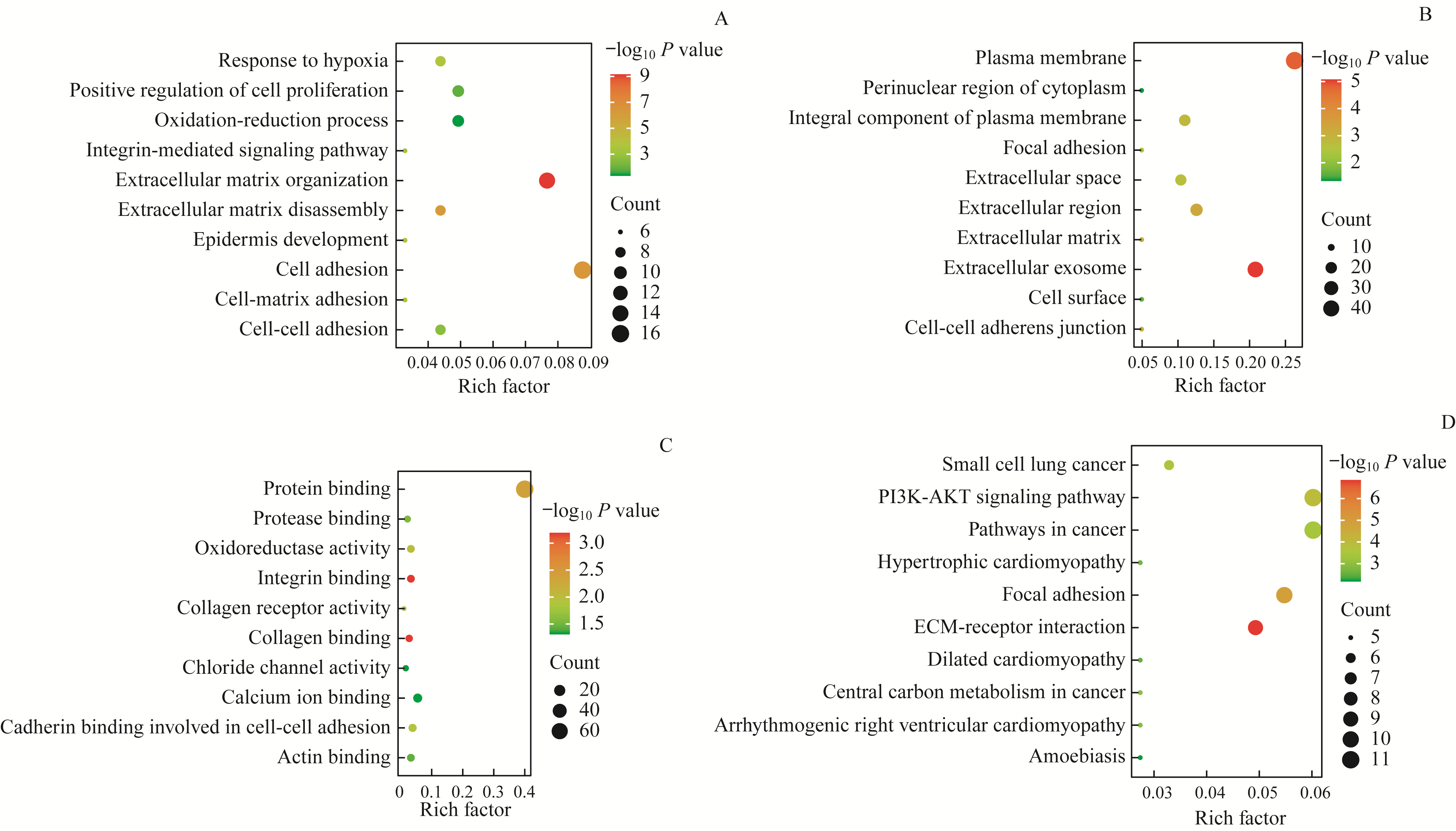
Fig 2 Enrichment analysis of GO function and KEGG pathway of DEGs

Fig 3 PPI network diagram of DEGs corresponding proteins
| Gene | Degree | Biological characteristic |
|---|---|---|
| FN1 | 24 | Involved in cell adhesion, cell movement, wound healing and maintenance of cell morphology |
| MET | 15 | Regulating cell proliferation, dispersion, proliferation and differentiation |
| LAMB3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| LAMA3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| ITGA3 | 11 | Involved in cell adhesion, invasion foot formation and matrix degradation |
Tab 1 Biological characteristics of the five DEGs in PPI network
| Gene | Degree | Biological characteristic |
|---|---|---|
| FN1 | 24 | Involved in cell adhesion, cell movement, wound healing and maintenance of cell morphology |
| MET | 15 | Regulating cell proliferation, dispersion, proliferation and differentiation |
| LAMB3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| LAMA3 | 11 | Involved in cell adhesion, migration and interaction with other extracellular matrix components; playing an important role in regulating cell migration and mechanical signal transduction |
| ITGA3 | 11 | Involved in cell adhesion, invasion foot formation and matrix degradation |
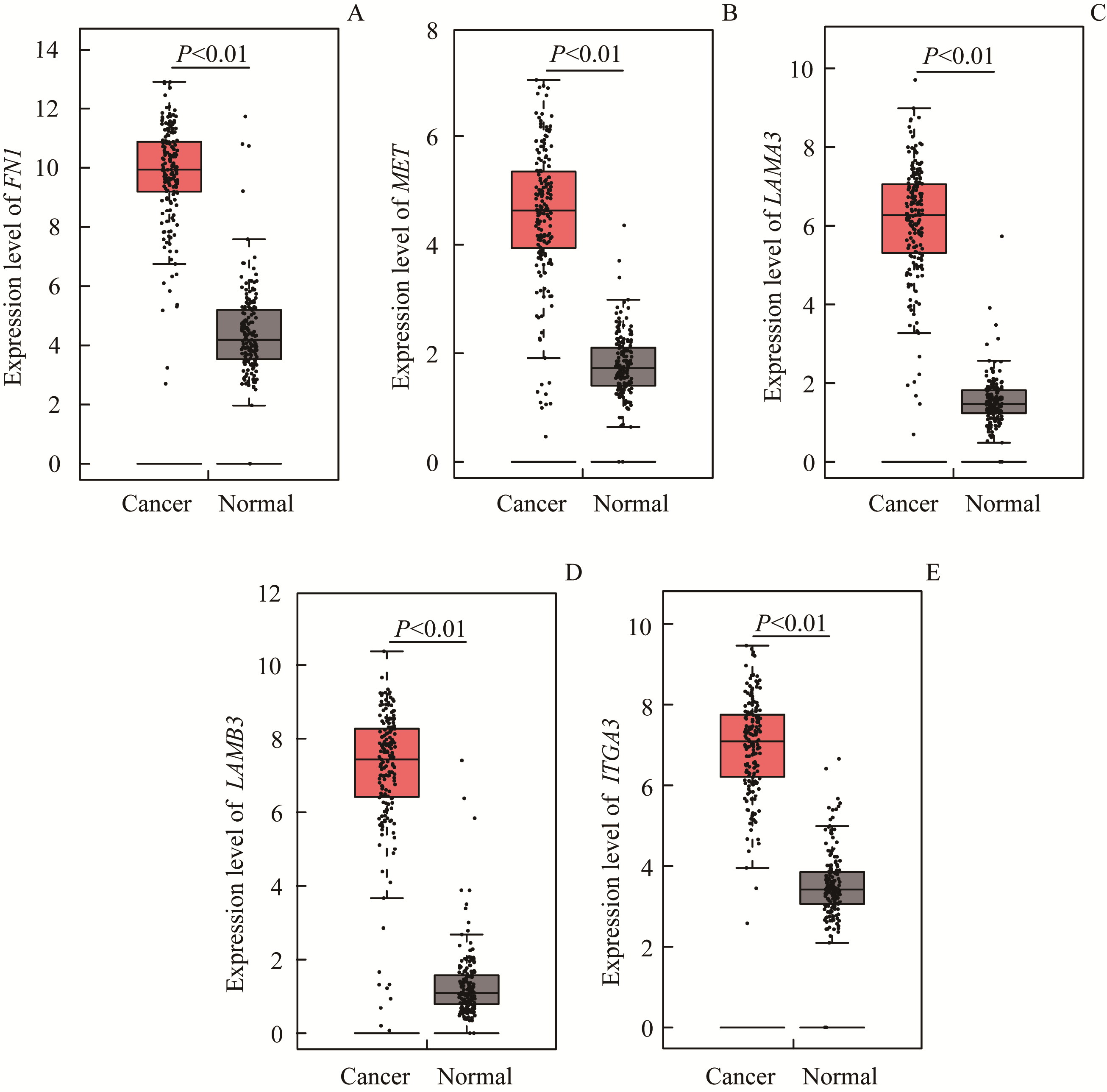
Fig 4 Expression of the five DEGs in 179 cases of pancreatic cancer tissues and 171 cases of adjacent normal tissues
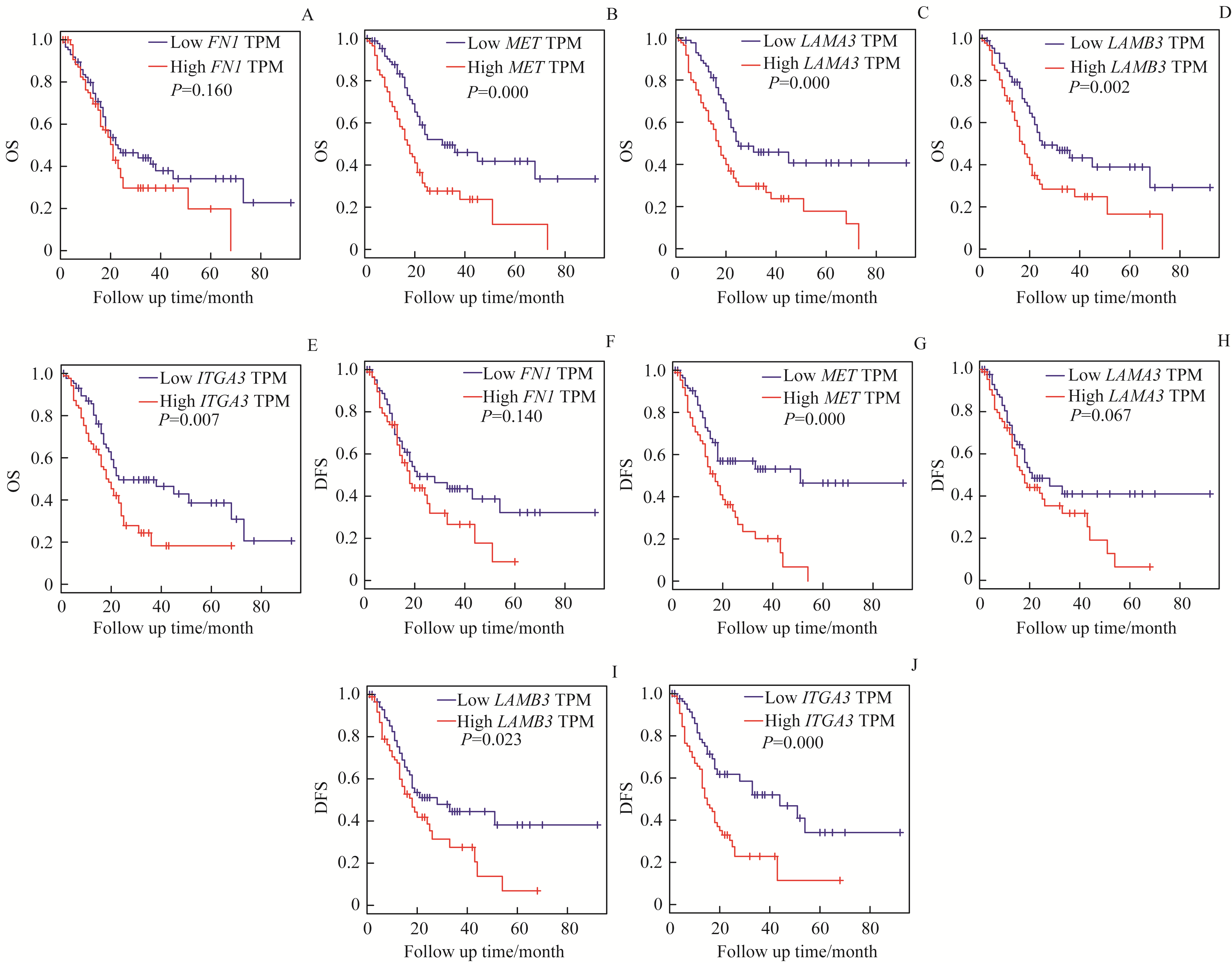
Fig 5 Prognostic significance of the five DEGs for pancreatic cancer patients assessed via Kaplan-Meier analysis
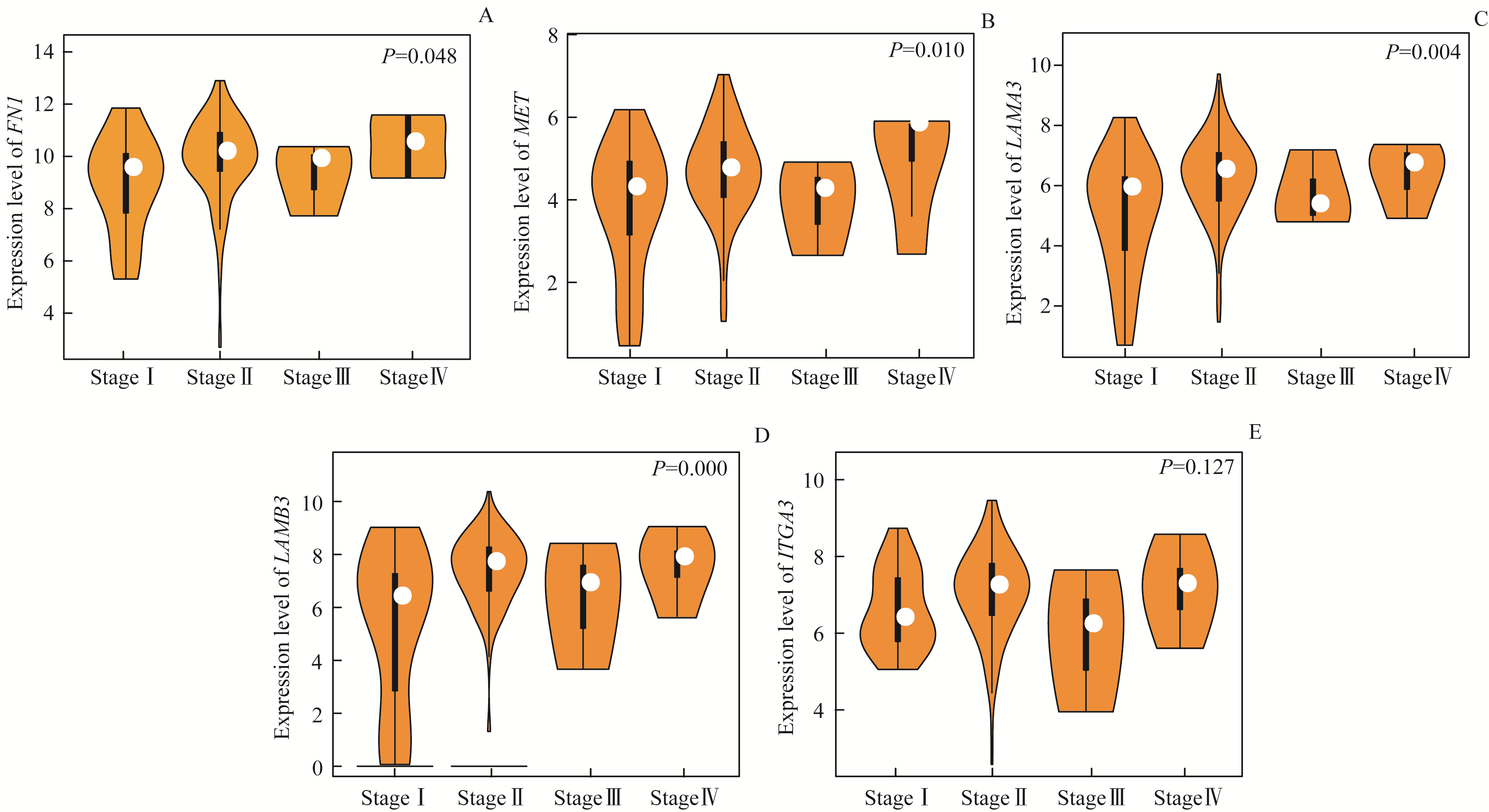
Fig 6 Expression of the five DEGs in pancreatic carcinoma with different pathological stages
| 1 | Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2018, 68(6): 394-424. |
| 2 | Ferlay J, Partensky C, Bray F. More deaths from pancreatic cancer than breast cancer in the EU by 2017[J]. Acta Oncol, 2016, 55(9/10): 1158-1160. |
| 3 | Lin QJ, Yang F, Jin C, et al. Current status and progress of pancreatic cancer in China[J]. World J Gastroenterol, 2015, 21(26): 7988-8003. |
| 4 | Mayo SC, Nathan H, Cameron JL, et al. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent[J]. Cancer, 2012, 118(10): 2674-2681. |
| 5 | de Sá MC, Simão ANC, de Medeiros FA, et al. Cell adhesion molecules and plasminogen activator inhibitor type-1 (PAI-1) in patients with rheumatoid arthritis: influence of metabolic syndrome[J]. Clin Exp Med, 2018, 18(4): 495-504. |
| 6 | Bendas G, Borsig L. Heparanase in cancer metastasis: heparin as a potential inhibitor of cell adhesion molecules[J]. Adv Exp Med Biol, 2020, 1221: 309-329. |
| 7 | Bergmann F, Wandschneider F, Sipos B, et al. Elevated L1CAM expression in precursor lesions and primary and metastastic tissues of pancreatic ductal adenocarcinoma[J]. Oncol Rep, 2010, 24(4): 909-915. |
| 8 | Geismann C, Morscheck M, Koch D, et al. Up-regulation of L1CAM in pancreatic duct cells is transforming growth factor β1- and slug-dependent: role in malignant transformation of pancreatic cancer[J]. Cancer Res, 2009, 69(10): 4517-4526. |
| 9 | Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment[J]. Annu Rev Med, 2016, 67: 11-28. |
| 10 | Liang H, Mokrani A, Chisomo-Kasiya H, et al. Dietary leucine affects glucose metabolism and lipogenesis involved in TOR/PI3K/Akt signaling pathway for juvenile blunt snout bream Megalobrama amblycephala[J]. Fish Physiol Biochem, 2019, 45(2): 719-732. |
| 11 | Polivka J, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway[J]. Pharmacol Ther, 2014, 142(2): 164-175. |
| 12 | Akinleye A, Avvaru P, Furqan M, et al. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics[J]. J Hematol Oncol, 2013, 6(1): 88. |
| 13 | Sierra JR, Tsao MS. C-MET as a potential therapeutic target and biomarker in cancer[J]. Ther Adv Med Oncol, 2011, 3(1): S21-S35. |
| 14 | Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification[J]. J Thorac Oncol, 2011, 6(5): 942-946. |
| 15 | Li Y, Chen CQ, He YL, et al. Abnormal expression of E-cadherin in tumor cells is associated with poor prognosis of gastric carcinoma[J]. J Surg Oncol, 2012, 106(3): 304-310. |
| 16 | di Renzo MF, Olivero M, Giacomini A, et al. Overexpression and amplification of themet/HGF receptor gene during the progression of colorectal cancer[J]. Clin Cancer Res, 1995, 1(2): 147-154. |
| 17 | Garcia S, Dalès JP, Jacquemier J, et al. C-Met overexpression in inflammatory breast carcinomas: automated quantification on tissue microarrays[J]. Br J Cancer, 2007, 96(2): 329-335. |
| 18 | Radaeva S, Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: a link between biliary intestinal Metaplasia and mucin-producing cholangiocarcinoma[J]. Hepatology, 1999, 29(5): 1453-1462. |
| 19 | di Renzo MF, Poulsom R, Olivero M, et al. Expression of the Met/hepatocyte growth factor receptor in human pancreatic cancer[J]. Cancer Res, 1995, 55(5): 1129-1138. |
| 20 | Neuzillet C, Couvelard A, Tijeras-Raballand A, et al. High c-Met expression in stage Ⅰ‒Ⅱ pancreatic adenocarcinoma: proposal for an immunostaining scoring method and correlation with poor prognosis[J]. Histopathology, 2015, 67(5): 664-676. |
| 21 | Hervieu A, Kermorgant S. The role of PI3K in Met driven cancer: a recap[J]. Front Mol Biosci, 2018, 5: 86. |
| 22 | Marinkovich MP. Tumour microenvironment: laminin 332 in squamous-cell carcinoma[J]. Nat Rev Cancer, 2007, 7(5): 370-380. |
| 23 | Stemmler S, Parwez Q, Petrasch-Parwez E, et al. Association of variation in the LAMA3 gene, encoding the α-chain of laminin 5, with atopic dermatitis in a German case-control cohort[J]. BMC Dermatol, 2014, 14: 17. |
| 24 | Castro BGR, Dos Reis R, Cintra GF, et al. Predictive factors for surgical morbidities and adjuvant chemotherapy delay for advanced ovarian cancer patients treated by primary debulking surgery or interval debulking surgery[J]. Int J Gynecol Cancer, 2018, 28(8): 1520-1528. |
| 25 | Lincoln V, Cogan J, Hou Y, et al. Gentamicin induces LAMB3 nonsense mutation readthrough and restores functional laminin 332 in junctional epidermolysis bullosa[J]. PNAS, 2018, 115(28): E6536-E6545. |
| 26 | Svoboda M, Hlobilová M, Marešová M, et al. Comparison of suction blistering and tape stripping for analysis of epidermal genes, proteins and lipids[J]. Arch Dermatol Res, 2017, 309(9): 757-765. |
| 27 | Wang YH, Jin YX, Bhandari A, et al. Upregulated LAMB3 increases proliferation and metastasis in thyroid cancer[J]. Onco Targets Ther, 2018, 11: 37-46. |
| 28 | Pan ZF, Li L, Fang QL, et al. Analysis of dynamic molecular networks for pancreatic ductal adenocarcinoma progression[J]. Cancer Cell Int, 2018, 18: 214. |
| 29 | Jung SN, Lim HS, Liu LH, et al. LAMB3 mediates metastatic tumor behavior in papillary thyroid cancer by regulating c-MET/Akt signals[J]. Sci Rep, 2018, 8(1): 2718. |
| 30 | Huang WJ, Gu JY, Tao T, et al. MiR-24-3p inhibits the progression of pancreatic ductal adenocarcinoma through LAMB3 downregulation[J]. Front Oncol, 2019, 9: 1499. |
| 31 | Zhang H, Pan YZ, Cheung M, et al. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway[J]. Cell Death Dis, 2019, 10(3): 230. |
| [1] | Wang Yiran, Zhang Zhe, Shen Jianfeng. Comprehensive analysis of the function, prognosis, and immune infiltration characteristics of SF3B1 mutations in uveal melanoma [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2026, 46(4): 475-485. |
| [2] | YU Zhiyuan, DONG Haiping, GAO Nan, MA Ke. Identification and mechanistic analysis of core genes associated with morphine tolerance in dorsal root ganglion: an integrative transcriptomics approach using WGCNA and machine learning algorithms [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(10): 1308-1319. |
| [3] | SUN Chenwei, HAI Wangxi, QU Qian, XI Yun. [18F]F-FMISO and [18F]F-FLT PET/CT dual-nuclide imaging for in vivo prediction of drug resistance in pancreatic cancer [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2025, 45(1): 60-68. |
| [4] | WEI Yunxin, JIANG Xushun, CAI Mengyao, WEN Ruizhi, DU Xiaogang. Correlation analysis of COMP and autophagy in diabetic nephropathy and its functional verification [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(7): 847-858. |
| [5] | WANG Xin, WANG Xiaoxia, LI Yanqing, ZHENG Yongxin, WU Jie, REN Meng, JIA Xiangdong, XU Tianxiang. Expression and clinical significance of geranylgeranyl diphosphate synthase1 (GGPS1) in lung squamous cell carcinoma [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(3): 312-324. |
| [6] | DENG Qingsong, ZHANG Changqing, TAO Shicong. Exploration of the relationship between nicotinamide metabolism-related genes and osteoarthritis [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(2): 145-160. |
| [7] | YU Siwei, XU Ziqi, TAO Mengyu, FAN Guangjian. Mechanistic study on the promotion of pancreatic cancer progression through upregulation of ZNF143 by dysregulated fatty acid metabolism [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2024, 44(10): 1255-1265. |
| [8] | DU Shaoqian, TAO Mengyu, CAO Yuan, WANG Hongxia, HU Xiaoqu, FAN Guangjian, ZANG Lijuan. CXCL9 expression in breast cancer and its correlation with the characteristics of tumor immunoinfiltration [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2023, 43(7): 860-872. |
| [9] | LI Qinglin, WANG Wenbo, LIU Wei. Bioinformatics analysis of pathological mechanism of degenerated tendon via stress deprivation [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2023, 43(5): 560-570. |
| [10] | QIN Yahan, ZHANG Ke, ZHANG Mengyu, SHEN Jie, PENG Meiyu. Research progress of MDSCs-targeted immunotherapy for pancreatic cancer [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2023, 43(10): 1317-1323. |
| [11] | MA Fangfang, QIN Jiejie, REN Lingjie, TANG Xiaomei, LIU Jia, SHI Minmin, JIANG Lingxi. Establishment of a 3D culture model in vitro of pancreatic cancer primary cells using hydrogel microspheres [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2023, 43(1): 79-87. |
| [12] | HAN Xiaxia, JIANG Yang, GU Shuangshuang, DAI Dai, SHEN Nan. Transcriptomic analysis of metabolic characteristics of the immune cells in systemic lupus erythematosus patients [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(9): 1197-1207. |
| [13] | JIN Bu, YUAN Ying, CHEN Wanyu, XU Hudong, HUANG Xiaolei, HE Jialu, YU Hong. Involvement of miRNA target genes in esophageal squamous cell carcinoma through ubiquitination based on GEO database [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(4): 464-471. |
| [14] | GONG Qiyu, CHEN Lei. Role of circulating and infiltrating B cells in immune microenvironment of colorectal cancer by multi-omics data profiling [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(4): 472-481. |
| [15] | XU Jingxuan, DU Shaoqian, CAO Yuan, WANG Hongxia, HUANG Weiyi. MMP14 expression in pancreatic cancer and its correlation with characteristics of tumor immune microenvironment [J]. Journal of Shanghai Jiao Tong University (Medical Science), 2022, 42(3): 312-322. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||