图1
基于表观转录靶向小分子药物库与功能基因组筛选GBM 和DIPG 共同的潜在药物靶标—— CDK12/13
Note : A. Workflow of epigenetic transcription-related targeted small molecule drug library screening in aGBM (U251), pGBM (SF188) and DIPG (DIPG_13F, DIPG_BJ02) cell lines. B. Venn diagram of the epigenetic transcription-related targeted small molecule drug library screening in U251, SF188, DIPG_13F, and DIPG_BJ02 cell lines. C. Heatmap was used to display the screening effects of 17 small molecule classifications. D. Scatter plots showing the dependence of GBM cells on CDK12 , CDK13 and CDK9 in DepMap database. E. Epigenetic transcription-related functional genome screening based on CRISPR-Cas9 in vitro for four weeks in DIPG_BJ02 cell line, with the sequencing results analyzed by MAGeCK algorithm. The top 200 genes are thought to be growth-dependent genes in DIPG_BJ02 cell line. F. Box plot of log (fold change) in sgCDK12 , sgCDK13 and sgCDK9 . NTC—negative control. ① P <0.000 1, ② P =0.001 8, ③ P =0.002 9.
Fig 1
Screening for common potential drug targets for GBM and DIPG based on epigenetic transcription-related targeted small molecule drug library and functional genome: CDK12 and CDK13
图2
CDK12 DIPG 和GBM 肿瘤细胞体外生长的抑制作用
Note : A. Western blotting analysis of CDK12 expression levels in aGBM (U251), pGBM (SF188, KNS42), and DIPG17 cell lines after CDK12 knockout by using CRISPR-Cas9 system. B. Detection of cell viability of U251, SF188 and KNS42 on the fourth day, and DIPG17 on the seventh day after CDK12 knockout by CellTiter-Glo. C. Dot plot (upper) and bar chart (below) of neurosphere assay of DIPG17 cells. Fifty or 100 DIPG17 cells were seeded in each well (96-well plate) following CDK12 knockout. After 14 d of culture, the number of neurosphere was counted and mapped. D. Scanogram (left) and cartogram (right) of colony formation assay of CDK12 -knockout GBM cell lines (U251, SF188 and KNS42). Cells were plated at 2 000 per well (six well plate), with half-volume mediumeing changed every 4 d, followed by crystal violet staining, photographing and counting in two weeks. ① P =0.000 2, ② P =0.000 4, ③ P =0.000 1, ④ P =0.008 7, ⑤ P =0.001 4, ⑥ P =0.004 0, ⑦ P =0.002 9, ⑧ P <0.000 1.
Fig 2
Inhibitory effect of CDK12 knockout on the growth of DIPG and GBM tumor cells in vitro
[1]
MILLER K D, OSTROM Q T, KRUCHKO C, et al. Brain and other central nervous system tumor statistics, 2021[J]. CA Cancer J Clin, 2021, 71(5): 381-406.
[本文引用: 1]
[2]
CHOI S, YU Y, GRIMMER M R, et al. Temozolomide-associated hypermutation in gliomas[J]. Neuro Oncol, 2018, 20(10): 1300-1309.
[本文引用: 1]
[3]
OSTROM Q T, PRICE M, NEFF C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015—2019[J]. Neuro Oncol, 2022, 24(Suppl 5): v1-v95.
[本文引用: 2]
[4]
MCKINNON C, NANDHABALAN M, MURRAY S A, et al. Glioblastoma: clinical presentation, diagnosis, and management[J]. BMJ, 2021, 374: n1560.
[本文引用: 2]
[5]
BRENNAN C W, VERHAAK R G, MCKENNA A, et al. The somatic genomic landscape of glioblastoma[J]. Cell, 2013, 155(2): 462-477.
[本文引用: 1]
[6]
MENG W, WANG J J, WANG B C, et al. CDK7 inhibition is a novel therapeutic strategy against GBM both in vitro and in vivo [J]. Cancer Manag Res, 2018, 10: 5747-5758.
[本文引用: 4]
[7]
YAN G, WANG Y F, CHEN J C, et al. Advances in drug development for targeted therapies for glioblastoma[J]. Med Res Rev, 2020, 40(5): 1950-1972.
[本文引用: 1]
[8]
NJONKOU R, JACKSON C M, WOODWORTH G F, et al. Pediatric glioblastoma: mechanisms of immune evasion and potential therapeutic opportunities[J]. Cancer Immunol Immunother, 2022, 71(8): 1813-1822.
[本文引用: 1]
[9]
BERGER T R, WEN P Y, LANG-ORSINI M, et al. World Health Organization 2021 classification of central nervous system tumors and implications for therapy for adult-type gliomas: a review[J]. JAMA Oncol, 2022, 8(10): 1493-1501.
[本文引用: 1]
[10]
ŚLEDZIŃSKA P, BEBYN M G, FURTAK J, et al. Prognostic and predictive biomarkers in gliomas[J]. Int J Mol Sci, 2021, 22(19): 10373.
[本文引用: 1]
[11]
PATHANIA M, DE JAY N, MAESTRO N, et al. H3.3K27M cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas[J]. Cancer Cell, 2017, 32(5): 684-700.e9.
[本文引用: 1]
[12]
HOFFMAN L M, VELDHUIJZEN VAN ZANTEN S E M, COLDITZ N, et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the international and European society for pediatric oncology DIPG registries[J]. J Clin Oncol, 2018, 36(19): 1963-1972.
[本文引用: 1]
[13]
AZIZ-BOSE R, MONJE M. Diffuse intrinsic pontine glioma: molecular landscape and emerging therapeutic targets[J]. Curr Opin Oncol, 2019, 31(6): 522-530.
[本文引用: 1]
[14]
RASHED W M, MAHER E, ADEL M, et al. Pediatric diffuse intrinsic pontine glioma: where do we stand?[J]. Cancer Metastasis Rev, 2019, 38(4): 759-770.
[本文引用: 2]
[15]
COONEY T M, LUBANSZKY E, PRASAD R, et al. Diffuse midline glioma: review of epigenetics[J]. J Neurooncol, 2020, 150(1): 27-34.
[本文引用: 1]
[16]
MENG W, WANG B C, MAO W W, et al. Enhanced efficacy of histone deacetylase inhibitor combined with bromodomain inhibitor in glioblastoma[J]. J Exp Clin Cancer Res, 2018, 37(1): 241.
[本文引用: 1]
[17]
MO J L, TAN K Z, DONG Y, et al. Therapeutic targeting the oncogenic driver EWSR1-FLI1 in Ewing sarcoma through inhibition of the FACT complex[J]. Oncogene, 2023, 42(1): 11-25.
[18]
NAGARAJA S, VITANZA N A, WOO P J, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma[J]. Cancer Cell, 2017, 31(5): 635-652.e6.
[本文引用: 2]
[19]
NGUYEN T T T, ZHANG Y R, SHANG E Y, et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models[J]. J Clin Invest, 2020, 130(7): 3699-3716.
[本文引用: 3]
[20]
ANASTAS J N, ZEE B M, KALIN J H, et al. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG[J]. Cancer Cell, 2019, 36(5): 528-544.e10.
[本文引用: 1]
[21]
DAHL N A, DANIS E, BALAKRISHNAN I, et al. Super elongation complex as a targetable dependency in diffuse midline glioma[J]. Cell Rep, 2020, 31(1): 107485.
[本文引用: 1]
[22]
RANJAN A, PANG Y, BUTLER M, et al. Targeting CDK9 for the treatment of glioblastoma[J]. Cancers (Basel), 2021, 13(12): 3039.
[本文引用: 3]
[23]
CHOU J, QUIGLEY D A, ROBINSON T M, et al. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy[J]. Cancer Discov, 2020, 10(3): 351-370.
[本文引用: 3]
[24]
LIANG K W, GAO X, GILMORE J M, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing[J]. Mol Cell Biol, 2015, 35(6): 928-938.
[本文引用: 1]
[25]
CHOI S H, KIM S, JONES K A. Gene expression regulation by CDK12: a versatile kinase in cancer with functions beyond CTD phosphorylation[J]. Exp Mol Med, 2020, 52(5): 762-771.
[本文引用: 1]
[26]
QUEREDA V, BAYLE S, VENA F, et al. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer[J]. Cancer Cell, 2019, 36(5): 545-558.e7.
[本文引用: 11]
[27]
WANG C, WANG H, LIEFTINK C, et al. CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma[J]. Gut, 2020, 69(4): 727-736.
[本文引用: 4]
[28]
DIETER S M, SIEGL C, CODÓ P L, et al. Degradation of CCNK/CDK12 is a druggable vulnerability of colorectal cancer[J]. Cell Rep, 2021, 36(3): 109394.
[本文引用: 1]
[29]
LIU H, SHIN S H, CHEN H Y, et al. CDK12 and PAK2 as novel therapeutic targets for human gastric cancer[J]. Theranostics, 2020, 10(14): 6201-6215.
[本文引用: 2]
[30]
JIANG B S, JIANG J, KALTHEUNER I H, et al. Structure-activity relationship study of THZ531 derivatives enables the discovery of BSJ-01-175 as a dual CDK12/13 covalent inhibitor with efficacy in Ewing sarcoma[J]. Eur J Med Chem, 2021, 221: 113481.
[本文引用: 2]
[31]
BLAZEK D, KOHOUTEK J, BARTHOLOMEEUSEN K, et al. The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes[J]. Genes Dev, 2011, 25(20): 2158-2172.
[本文引用: 1]
[32]
DUBBURY S J, BOUTZ P L, SHARP P A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation[J]. Nature, 2018, 564(7734): 141-145.
[本文引用: 1]
[33]
ZHANG T H, KWIATKOWSKI N, OLSON C M, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors[J]. Nat Chem Biol, 2016, 12(10): 876-884.
[本文引用: 3]
[34]
FENG J X, MEYER C A, WANG Q, et al. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data[J]. Bioinformatics, 2012, 28(21): 2782-2788.
[本文引用: 1]
[35]
INIGUEZ A B, STOLTE B, WANG E J, et al. EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in Ewing sarcoma[J]. Cancer Cell, 2018, 33(2): 202-216.e6.
[本文引用: 3]
[36]
COLLINS P L, PURMAN C, PORTER S I, et al. DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner[J]. Nat Commun, 2020, 11(1): 3158.
[本文引用: 1]
[37]
LI R, OKADA H, YAMASHITA T, et al. FOXM1 is a novel molecular target of AFP-positive hepatocellular carcinoma abrogated by proteasome inhibition[J]. Int J Mol Sci, 2022, 23(15): 8305.
[本文引用: 2]
[38]
HOPKINS J L, ZOU L. Induction of BRCAness in triple-negative breast cancer by a CDK12/13 inhibitor improves chemotherapy[J]. Cancer Cell, 2019, 36(5): 461-463.
[本文引用: 1]
[39]
NIU T, LI K L, JIANG L, et al. Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-cyclin K complex and induce synthetic lethality with PARP inhibitor[J]. Eur J Med Chem, 2022, 228: 114012.
[本文引用: 1]
[40]
YAM C Q X, LIM H H, SURANA U. DNA damage checkpoint execution and the rules of its disengagement[J]. Front Cell Dev Biol, 2022, 10: 1020643.
[本文引用: 1]
[41]
XIE J B, SHEN Z Y, ANRAKU Y, et al. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies[J]. Biomaterials, 2019, 224: 119491.
[本文引用: 2]
[42]
BOBO R H, LASKE D W, AKBASAK A, et al. Convection-enhanced delivery of macromolecules in the brain[J]. Proc Natl Acad Sci U S A, 1994, 91(6): 2076-2080.
[本文引用: 1]
[43]
MADANI F, LINDBERG S, LANGEL U, et al. Mechanisms of cellular uptake of cell-penetrating peptides[J]. J Biophys, 2011, 2011: 414729.
[本文引用: 1]
[44]
HERAVI SHARGH V, LUCKETT J, BOUZINAB K, et al. Chemosensitization of temozolomide-resistant pediatric diffuse midline glioma using potent nanoencapsulated forms of a N(3)-propargyl analogue[J]. ACS Appl Mater Interfaces, 2021, 13(30): 35266-35280.
[本文引用: 1]
[45]
WU T T, LIU Y, CAO Y, et al. Engineering macrophage exosome disguised biodegradable nanoplatform for enhanced sonodynamic therapy of glioblastoma[J]. Adv Mater, 2022, 34(15): e2110364.
[本文引用: 1]
1
... 脑和其他中枢神经系统肿瘤是致死率最高的肿瘤类型之一,其中恶性肿瘤占比不到30%,却是导致死亡的主要原因.这些恶性肿瘤中胶质瘤占比最多,约80%[1 ] .世界卫生组织(World Health Organization,WHO)将胶质瘤按侵袭性分为4个组织学程度(WHO Ⅰ~Ⅳ级),其中WHO Ⅲ和WHO Ⅳ级胶质瘤被称为高级别胶质瘤(high-grade gliomas,HGGs)[2 ] .WHO Ⅳ级的HGGs主要包括胶质母细胞瘤(glioblastoma,GBM)和弥漫性内生性脑桥胶质瘤(diffuse intrinsic pontine glioma,DIPG)2个类型[3 ] . ...
1
... 脑和其他中枢神经系统肿瘤是致死率最高的肿瘤类型之一,其中恶性肿瘤占比不到30%,却是导致死亡的主要原因.这些恶性肿瘤中胶质瘤占比最多,约80%[1 ] .世界卫生组织(World Health Organization,WHO)将胶质瘤按侵袭性分为4个组织学程度(WHO Ⅰ~Ⅳ级),其中WHO Ⅲ和WHO Ⅳ级胶质瘤被称为高级别胶质瘤(high-grade gliomas,HGGs)[2 ] .WHO Ⅳ级的HGGs主要包括胶质母细胞瘤(glioblastoma,GBM)和弥漫性内生性脑桥胶质瘤(diffuse intrinsic pontine glioma,DIPG)2个类型[3 ] . ...
2
... 脑和其他中枢神经系统肿瘤是致死率最高的肿瘤类型之一,其中恶性肿瘤占比不到30%,却是导致死亡的主要原因.这些恶性肿瘤中胶质瘤占比最多,约80%[1 ] .世界卫生组织(World Health Organization,WHO)将胶质瘤按侵袭性分为4个组织学程度(WHO Ⅰ~Ⅳ级),其中WHO Ⅲ和WHO Ⅳ级胶质瘤被称为高级别胶质瘤(high-grade gliomas,HGGs)[2 ] .WHO Ⅳ级的HGGs主要包括胶质母细胞瘤(glioblastoma,GBM)和弥漫性内生性脑桥胶质瘤(diffuse intrinsic pontine glioma,DIPG)2个类型[3 ] . ...
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
2
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
... GBM和DIPG这2类HGGs是恶性程度最高、患者预后最差的胶质瘤类型,目前最大限度地手术并结合放化疗是针对它们的主要治疗方案[4 ,14 ] ,但其非特异损伤较大,患者预后差,亟需寻找精准有效的靶向治疗方法.近年来,大量研究表明表观转录调控因子是癌症的关键依赖基因和有效的治疗靶点.表观转录靶向策略在多种癌症研究模型中呈现出显著的治疗效果,多类表观转录靶向小分子药物进入肿瘤治疗的人体临床试验.鉴于DIPG和GBM中被报道过很多相同的表观转录治疗靶标与靶向策略[6 ,19 -22 ] ,本研究计划通过对多个DIPG和GBM细胞系进行针对表观转录调控因子的功能基因组与靶向小分子药物库筛选分析,系统鉴定二者共同的表观转录治疗新靶点和新策略,并进一步进行体外功能验证与机制挖掘,以期为HGGs的靶向治疗提供参考. ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
4
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... GBM和DIPG这2类HGGs是恶性程度最高、患者预后最差的胶质瘤类型,目前最大限度地手术并结合放化疗是针对它们的主要治疗方案[4 ,14 ] ,但其非特异损伤较大,患者预后差,亟需寻找精准有效的靶向治疗方法.近年来,大量研究表明表观转录调控因子是癌症的关键依赖基因和有效的治疗靶点.表观转录靶向策略在多种癌症研究模型中呈现出显著的治疗效果,多类表观转录靶向小分子药物进入肿瘤治疗的人体临床试验.鉴于DIPG和GBM中被报道过很多相同的表观转录治疗靶标与靶向策略[6 ,19 -22 ] ,本研究计划通过对多个DIPG和GBM细胞系进行针对表观转录调控因子的功能基因组与靶向小分子药物库筛选分析,系统鉴定二者共同的表观转录治疗新靶点和新策略,并进一步进行体外功能验证与机制挖掘,以期为HGGs的靶向治疗提供参考. ...
... 筛选结果显示在本研究所测试的2个DIPG和2个GBM细胞系中均有显著抑制作用的表观转录靶向小分子共17个,多为HDAC和CDK的抑制剂,并且其中大部分在DIPG或GBM中已经有过报道[6 ,19 -22 ] ,证明了筛选的可信度高.在CDK抑制剂中,本研究重点关注到了CDK12/13靶向小分子THZ531[33 ] 尚未在DIPG和GBM中被报道过.而在针对DIPG和GBM对CDK12/13 生长依赖性的评估中,DepMap数据库中GBM细胞系的依赖性分析数据以及利用DIPG_BJ02细胞系进行的表观转录靶向的CRISPR-Cas9功能基因组筛选结果,均表明GBM和DIPG细胞对CDK12/13 有显著依赖性(图1 ).在此基础上,进一步利用多个GBM和DIPG细胞系验证了无论是用基于CRISPR-Cas9的遗传学方法或是2个不同的靶向CDK12/13的小分子抑制剂(THZ531和SR-4835)均能产生显著的体外肿瘤抑制效应(图2 和图3 ).以上结果均表明CDK12/13 是很有潜力的HGGs治疗新靶标. ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
2
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
... GBM和DIPG这2类HGGs是恶性程度最高、患者预后最差的胶质瘤类型,目前最大限度地手术并结合放化疗是针对它们的主要治疗方案[4 ,14 ] ,但其非特异损伤较大,患者预后差,亟需寻找精准有效的靶向治疗方法.近年来,大量研究表明表观转录调控因子是癌症的关键依赖基因和有效的治疗靶点.表观转录靶向策略在多种癌症研究模型中呈现出显著的治疗效果,多类表观转录靶向小分子药物进入肿瘤治疗的人体临床试验.鉴于DIPG和GBM中被报道过很多相同的表观转录治疗靶标与靶向策略[6 ,19 -22 ] ,本研究计划通过对多个DIPG和GBM细胞系进行针对表观转录调控因子的功能基因组与靶向小分子药物库筛选分析,系统鉴定二者共同的表观转录治疗新靶点和新策略,并进一步进行体外功能验证与机制挖掘,以期为HGGs的靶向治疗提供参考. ...
1
... GBM分为成人GBM(adult GBM,aGBM)和儿童GBM(pediatric GBM,pGBM).目前对于GBM的治疗手段主要是最大限度地外科手术结合放射治疗(放疗)或者替莫唑胺(temozolomide,TMZ)治疗,但是几乎都会复发[4 ] .aGBM占恶性脑肿瘤的50.1%,是较常见的成人恶性原发性脑肿瘤,发病率随年龄增长而增加;75~84岁的老年人发病率最高,aGBM患者的5年生存率约为5%[3 ] .癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库分析结果表明aGBM中有3个核心通路的基因改变[5 ] :磷脂酰肌醇3激酶(PI3K)通路、p53通路和Rb通路.靶向3个核心通路是治疗aGBM的合理策略.此外,基因转录调控异常在癌症发生发展中起到重要作用,靶向转录辅因子是治疗癌症的有效手段,如周期蛋白依赖性激酶7(cyclin dependent kinase 7,CDK7)抑制剂THZ1是治疗aGBM的有效分子[6 ] .再者,也可以通过靶向肿瘤血管生成和免疫检查点途径治疗aGBM[7 ] .pGBM与aGBM相比较为罕见,占儿童脑肿瘤的15%[8 ] .2类肿瘤在组织学上相似,但是在生物学和分子特征上有着很大的不同[9 ] .约1/3的pGBM患者中编码组蛋白H3.3的基因H3F3A 具有多种突变[10 ] :第27位赖氨酸突变为甲硫氨酸(K27M),第34位甘氨酸突变为精氨酸(G34R)或缬氨酸(G34V).H3F3A 突变可以诱导pGBM过度增殖,但要结合其他驱动因素才能发展为恶性肿瘤[11 ] .另一类儿童HGGs是DIPG,好发于脑干的脑桥区域,中位生存期约为9个月,5年生存率不到1%[12 -13 ] .因DIPG发病位置特殊,且弥漫性生长,所以无法进行外科手术,只能采取放疗或者化学治疗(化疗)[14 ] .80%的DIPG患者存在H3K27M 突变,该突变在DIPG中同样发挥了重要的致癌作用[15 ] . ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
2
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... ,18 ]、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
3
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... GBM和DIPG这2类HGGs是恶性程度最高、患者预后最差的胶质瘤类型,目前最大限度地手术并结合放化疗是针对它们的主要治疗方案[4 ,14 ] ,但其非特异损伤较大,患者预后差,亟需寻找精准有效的靶向治疗方法.近年来,大量研究表明表观转录调控因子是癌症的关键依赖基因和有效的治疗靶点.表观转录靶向策略在多种癌症研究模型中呈现出显著的治疗效果,多类表观转录靶向小分子药物进入肿瘤治疗的人体临床试验.鉴于DIPG和GBM中被报道过很多相同的表观转录治疗靶标与靶向策略[6 ,19 -22 ] ,本研究计划通过对多个DIPG和GBM细胞系进行针对表观转录调控因子的功能基因组与靶向小分子药物库筛选分析,系统鉴定二者共同的表观转录治疗新靶点和新策略,并进一步进行体外功能验证与机制挖掘,以期为HGGs的靶向治疗提供参考. ...
... 筛选结果显示在本研究所测试的2个DIPG和2个GBM细胞系中均有显著抑制作用的表观转录靶向小分子共17个,多为HDAC和CDK的抑制剂,并且其中大部分在DIPG或GBM中已经有过报道[6 ,19 -22 ] ,证明了筛选的可信度高.在CDK抑制剂中,本研究重点关注到了CDK12/13靶向小分子THZ531[33 ] 尚未在DIPG和GBM中被报道过.而在针对DIPG和GBM对CDK12/13 生长依赖性的评估中,DepMap数据库中GBM细胞系的依赖性分析数据以及利用DIPG_BJ02细胞系进行的表观转录靶向的CRISPR-Cas9功能基因组筛选结果,均表明GBM和DIPG细胞对CDK12/13 有显著依赖性(图1 ).在此基础上,进一步利用多个GBM和DIPG细胞系验证了无论是用基于CRISPR-Cas9的遗传学方法或是2个不同的靶向CDK12/13的小分子抑制剂(THZ531和SR-4835)均能产生显著的体外肿瘤抑制效应(图2 和图3 ).以上结果均表明CDK12/13 是很有潜力的HGGs治疗新靶标. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
3
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... GBM和DIPG这2类HGGs是恶性程度最高、患者预后最差的胶质瘤类型,目前最大限度地手术并结合放化疗是针对它们的主要治疗方案[4 ,14 ] ,但其非特异损伤较大,患者预后差,亟需寻找精准有效的靶向治疗方法.近年来,大量研究表明表观转录调控因子是癌症的关键依赖基因和有效的治疗靶点.表观转录靶向策略在多种癌症研究模型中呈现出显著的治疗效果,多类表观转录靶向小分子药物进入肿瘤治疗的人体临床试验.鉴于DIPG和GBM中被报道过很多相同的表观转录治疗靶标与靶向策略[6 ,19 -22 ] ,本研究计划通过对多个DIPG和GBM细胞系进行针对表观转录调控因子的功能基因组与靶向小分子药物库筛选分析,系统鉴定二者共同的表观转录治疗新靶点和新策略,并进一步进行体外功能验证与机制挖掘,以期为HGGs的靶向治疗提供参考. ...
... 筛选结果显示在本研究所测试的2个DIPG和2个GBM细胞系中均有显著抑制作用的表观转录靶向小分子共17个,多为HDAC和CDK的抑制剂,并且其中大部分在DIPG或GBM中已经有过报道[6 ,19 -22 ] ,证明了筛选的可信度高.在CDK抑制剂中,本研究重点关注到了CDK12/13靶向小分子THZ531[33 ] 尚未在DIPG和GBM中被报道过.而在针对DIPG和GBM对CDK12/13 生长依赖性的评估中,DepMap数据库中GBM细胞系的依赖性分析数据以及利用DIPG_BJ02细胞系进行的表观转录靶向的CRISPR-Cas9功能基因组筛选结果,均表明GBM和DIPG细胞对CDK12/13 有显著依赖性(图1 ).在此基础上,进一步利用多个GBM和DIPG细胞系验证了无论是用基于CRISPR-Cas9的遗传学方法或是2个不同的靶向CDK12/13的小分子抑制剂(THZ531和SR-4835)均能产生显著的体外肿瘤抑制效应(图2 和图3 ).以上结果均表明CDK12/13 是很有潜力的HGGs治疗新靶标. ...
3
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... [23 ,25 ].靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... 已有研究[23 ] 表明CDK12/13 之间既有功能上的相互配合,也存在一定的冗余.基于以上的肿瘤依赖性分析结果(图1 D~F)和已有的CDK12/13 肿瘤依赖性相关报道[26 ] ,推测CDK12 可能相较于CDK13 来说发挥更重要的生长调控作用,因此挑选CDK12 进行肿瘤依赖性的独立验证.发现当利用CRISPR-Cas9方法在DIPG17、aGBM细胞系U251和pGBM细胞系(SF188、KNS42)中敲除CDK12 ,可以显著抑制上述HGGs细胞系的体外细胞活性(图2 A、B).在DIPG17细胞系中,成球实验结果显示敲除CDK12 可以显著抑制DIPG17细胞的自我更新能力(图2 C).而在另外3个GBM细胞系(U251、SF188、KNS42)中,敲除CDK12 也可显著抑制其克隆形成能力(图2 D).上述结果均表明CDK12 缺失能够显著抑制DIPG和GBM细胞的体外生长,验证了该基因的肿瘤依赖性. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
11
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... [26 ].研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... [26 ];其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... [26 ],说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... [26 ],均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... 为了系统寻找DIPG和GBM中共同的表观转录相关的靶向治疗新策略,在aGBM细胞系U251、pGBM细胞系SF188和2个DIPG细胞系(DIPG_13F、DIPG_BJ02)中进行表观转录相关的靶向小分子药物库筛选(图1 A).以1 µmol/L药物处理时抑制率大于50%作为筛选标准,分别有32、64、44和37个靶向小分子能够有效抑制U251、SF188、DIPG_13F和DIPG_BJ02细胞系生长,其中17个靶向小分子对4个细胞系均有显著抑制效果(图1 B).它们主要是HDAC抑制剂、CDK抑制剂和微管相关蛋白抑制剂(图1 C).关注到小分子抑制剂THZ531对应的CDK12/13靶向治疗虽然在三阴性乳腺癌、尤文肉瘤、胃癌、结直肠癌等多种肿瘤中被发现有效[26 -30 ] ,但在DIPG和GBM中尚未有过报道,因此接下来选择该类表观转录靶向治疗策略继续研究. ...
... 已有研究[23 ] 表明CDK12/13 之间既有功能上的相互配合,也存在一定的冗余.基于以上的肿瘤依赖性分析结果(图1 D~F)和已有的CDK12/13 肿瘤依赖性相关报道[26 ] ,推测CDK12 可能相较于CDK13 来说发挥更重要的生长调控作用,因此挑选CDK12 进行肿瘤依赖性的独立验证.发现当利用CRISPR-Cas9方法在DIPG17、aGBM细胞系U251和pGBM细胞系(SF188、KNS42)中敲除CDK12 ,可以显著抑制上述HGGs细胞系的体外细胞活性(图2 A、B).在DIPG17细胞系中,成球实验结果显示敲除CDK12 可以显著抑制DIPG17细胞的自我更新能力(图2 C).而在另外3个GBM细胞系(U251、SF188、KNS42)中,敲除CDK12 也可显著抑制其克隆形成能力(图2 D).上述结果均表明CDK12 缺失能够显著抑制DIPG和GBM细胞的体外生长,验证了该基因的肿瘤依赖性. ...
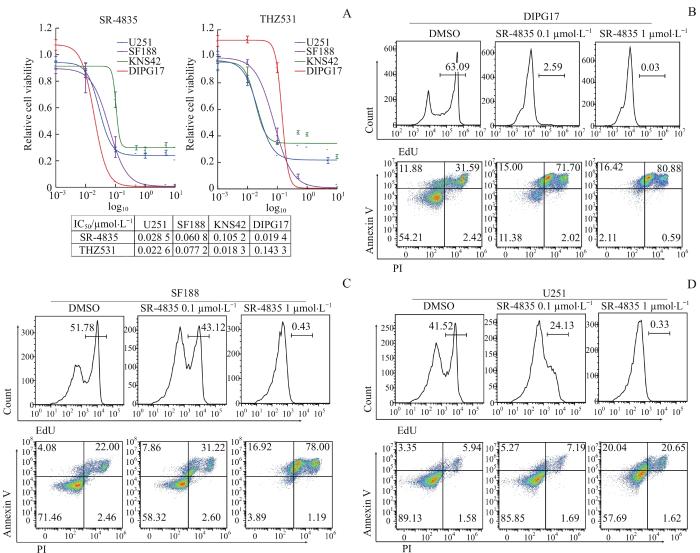
... 接下来,进一步分析CDK12/13抑制剂对GBM和DIPG的体外治疗作用.如图3 A所示,THZ531[33 ] 和SR-4835[26 ] 这2个高选择性的CDK12/13抑制剂对DIPG细胞系DIPG17以及GBM细胞系U251、SF188和KNS42均表现出剂量依赖性的细胞活性抑制作用,IC50 均小于1 µmol/L,与已报道的其他类型肿瘤细胞的结果相似[26 -27 ,35 ] .流式细胞术检测靶向抑制CDK12/13对DIPG和GBM细胞增殖和凋亡的影响,结果显示0.1 µmol/L或1 µmol/L的SR-4835均可以显著抑制DIPG17、SF188和U251细胞增殖和促进其细胞凋亡(图3 B~D). ...
... [26 -27 ,35 ].流式细胞术检测靶向抑制CDK12/13对DIPG和GBM细胞增殖和凋亡的影响,结果显示0.1 µmol/L或1 µmol/L的SR-4835均可以显著抑制DIPG17、SF188和U251细胞增殖和促进其细胞凋亡(图3 B~D). ...
... 根据药物剂量杀伤曲线结果(图3 A),选择0.1 µmol/L和1 µmol/L的SR-4835分别处理DIPG17细胞或SF188细胞20 h后的样本进行RNA-seq转录组测序,并重点对4个样本中相较于DMSO处理组表达水平显著下调的基因进行比较分析.如图4 A所示,与其他3个样本相比,0.1 µmol/L SR-4835处理的SF188细胞中表达水平显著下调的基因数量偏低,这与其细胞活性杀伤水平偏低相符合.因此,后续分析主要选择其他3个样本中表达水平显著下调基因的交集部分共994个基因作为研究对象.GO分析结果表明,这些基因显著富集的生物学过程与已知的CDK12/13参与调控的基因转录和DDR相关(图4 B).GSVA分析结果也显示,SR-4835能够显著下调DIPG17和SF188细胞中这2类生物学过程相关基因集合的转录水平,说明CDK12/13抑制剂作用于DIPG和GBM细胞时具有在靶(on-target)效应.已有研究[26 -27 ,35 ] 表明,在多个肿瘤类型中CDK12/13抑制剂能够通过显著拮抗DDR与放化疗以及其他靶向治疗手段之间产生显著的协同治疗效果.因此,对DIPG和GBM中靶向抑制CDK12/13是否能引起DDR相关基因的转录下调以及DNA损伤的积累进行进一步验证.如图4 D和5 A所示,通过RT-qPCR验证了SR-4835和THZ531能够在多个DIPG和GBM细胞系中引起系列关键DDR相关基因(TP53 、CCNK 、ATM 、ATR 、E2F1 、BRCA1 、FANCD2 、SMARCC2 、RAD51 、FANCI )的转录下调.此外,通过蛋白质印迹法(Western blotting)验证了SR-4835和THZ531处理能够显著下调DIPG和GBM细胞中RNA Pol Ⅱ CTD S2位点的磷酸化(RNA Pol Ⅱ S2P)水平,并且显著上调DNA双链断裂(double-strand break,DSB)标志物γH2AX(组蛋白H2AX第139位丝氨酸的磷酸化[36 ] )的水平,表明在这2类HGGs中靶向抑制CDK12/13确实能够引起DNA损伤的积累(图5 B). ...
... 为了探究HGGs中靶向抑制CDK12/13 拮抗肿瘤的分子机制,对SR-4835处理的DIPG细胞系DIPG17和pGBM细胞系SF188的RNA-seq结果进行分析,重点关注在2个细胞系中均表达下调的基因(Gfold<-1).GO分析表明,这些基因显著富集于细胞周期、基因转录、RNA剪切以及DNA修复等已知的CDK12/13参与调控的多个生物学过程或功能;进一步的GSVA分析也证明了以上生物学过程或功能受到CDK12/13抑制剂的拮抗;再结合在SR-4835和THZ531处理的多个GBM和DIPG细胞系中所检测到的RNA Pol Ⅱ S2P水平下降,均说明研究所使用的CDK12/13靶向小分子抑制剂发挥了on-target的抑制作用.在此基础上,本研究首先针对CDK12/13抑制剂引起的DNA损伤修复功能失调进行了验证.已有研究[26 ] 发现肿瘤细胞中的多个DNA损伤修复基因对CDK12/13抑制剂引起的转录下调非常敏感,这与本研究在多个HGGs细胞系中的RT-qPCR验证结果一致(图5 A).同时,检测到了双链DNA损伤标志物γH2AX蛋白磷酸化水平在CDK12/13抑制剂处理之后发生上调(图5 B),说明确实发生了DNA损伤的累积.GSVA分析结果还表明CDK12/13抑制剂主要下调双链DDR和同源重组修复相关基因(图4 C).以上结果均提示在GBM和DIPG中,CDK12/13抑制剂可能也像以往在众多肿瘤模型中报道[38 -39 ] 过的那样,能够与放化疗或者PARPi产生协同抑制效应或合成致死效应.此外,已有研究[40 ] 表明当DNA损伤不能得到及时修复时,细胞将启动G2-M期DNA损伤检查点,防止损伤细胞进入有丝分裂,这与检测到的CDK12/13抑制剂可引起DIPG和GBM细胞发生G2-M期细胞周期阻滞的结果相符合(图6 C).同时,RNA-seq分析还发现了CDK12/13抑制剂可引起蛋白酶体介导的泛素化蛋白降解过程相关基因的表达显著下调(图6 A).而根据以往报道[37 ] ,靶向抑制蛋白酶体功能同样可以使肿瘤细胞发生G2-M期阻滞.综上所述,本研究推测抑制CDK12/13导致DDR下调和蛋白酶体介导的泛素化蛋白降解减少很可能共同构成细胞G2-M期阻滞的上游分子事件.另一方面,本研究注意到GBM和DIPG细胞受CDK12抑制剂处理后各自存在很多特有的表达显著下调基因,这提示不同的HGGs细胞可能对CDK12抑制剂具有一些差异性的反应(图4 A).因此,进一步挖掘不同肿瘤类型特有的CDK12靶向效应及相关分子机制将有望为该类药物的临床应用提供更精准的指导和参考. ...
4
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... 接下来,进一步分析CDK12/13抑制剂对GBM和DIPG的体外治疗作用.如图3 A所示,THZ531[33 ] 和SR-4835[26 ] 这2个高选择性的CDK12/13抑制剂对DIPG细胞系DIPG17以及GBM细胞系U251、SF188和KNS42均表现出剂量依赖性的细胞活性抑制作用,IC50 均小于1 µmol/L,与已报道的其他类型肿瘤细胞的结果相似[26 -27 ,35 ] .流式细胞术检测靶向抑制CDK12/13对DIPG和GBM细胞增殖和凋亡的影响,结果显示0.1 µmol/L或1 µmol/L的SR-4835均可以显著抑制DIPG17、SF188和U251细胞增殖和促进其细胞凋亡(图3 B~D). ...
... 根据药物剂量杀伤曲线结果(图3 A),选择0.1 µmol/L和1 µmol/L的SR-4835分别处理DIPG17细胞或SF188细胞20 h后的样本进行RNA-seq转录组测序,并重点对4个样本中相较于DMSO处理组表达水平显著下调的基因进行比较分析.如图4 A所示,与其他3个样本相比,0.1 µmol/L SR-4835处理的SF188细胞中表达水平显著下调的基因数量偏低,这与其细胞活性杀伤水平偏低相符合.因此,后续分析主要选择其他3个样本中表达水平显著下调基因的交集部分共994个基因作为研究对象.GO分析结果表明,这些基因显著富集的生物学过程与已知的CDK12/13参与调控的基因转录和DDR相关(图4 B).GSVA分析结果也显示,SR-4835能够显著下调DIPG17和SF188细胞中这2类生物学过程相关基因集合的转录水平,说明CDK12/13抑制剂作用于DIPG和GBM细胞时具有在靶(on-target)效应.已有研究[26 -27 ,35 ] 表明,在多个肿瘤类型中CDK12/13抑制剂能够通过显著拮抗DDR与放化疗以及其他靶向治疗手段之间产生显著的协同治疗效果.因此,对DIPG和GBM中靶向抑制CDK12/13是否能引起DDR相关基因的转录下调以及DNA损伤的积累进行进一步验证.如图4 D和5 A所示,通过RT-qPCR验证了SR-4835和THZ531能够在多个DIPG和GBM细胞系中引起系列关键DDR相关基因(TP53 、CCNK 、ATM 、ATR 、E2F1 、BRCA1 、FANCD2 、SMARCC2 、RAD51 、FANCI )的转录下调.此外,通过蛋白质印迹法(Western blotting)验证了SR-4835和THZ531处理能够显著下调DIPG和GBM细胞中RNA Pol Ⅱ CTD S2位点的磷酸化(RNA Pol Ⅱ S2P)水平,并且显著上调DNA双链断裂(double-strand break,DSB)标志物γH2AX(组蛋白H2AX第139位丝氨酸的磷酸化[36 ] )的水平,表明在这2类HGGs中靶向抑制CDK12/13确实能够引起DNA损伤的积累(图5 B). ...
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
2
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
2
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... 为了系统寻找DIPG和GBM中共同的表观转录相关的靶向治疗新策略,在aGBM细胞系U251、pGBM细胞系SF188和2个DIPG细胞系(DIPG_13F、DIPG_BJ02)中进行表观转录相关的靶向小分子药物库筛选(图1 A).以1 µmol/L药物处理时抑制率大于50%作为筛选标准,分别有32、64、44和37个靶向小分子能够有效抑制U251、SF188、DIPG_13F和DIPG_BJ02细胞系生长,其中17个靶向小分子对4个细胞系均有显著抑制效果(图1 B).它们主要是HDAC抑制剂、CDK抑制剂和微管相关蛋白抑制剂(图1 C).关注到小分子抑制剂THZ531对应的CDK12/13靶向治疗虽然在三阴性乳腺癌、尤文肉瘤、胃癌、结直肠癌等多种肿瘤中被发现有效[26 -30 ] ,但在DIPG和GBM中尚未有过报道,因此接下来选择该类表观转录靶向治疗策略继续研究. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
1
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
3
... 表观转录调控分子已被揭示为癌症的关键调控因子和有效的治疗靶点.单独应用表观转录靶向治疗或联合其他治疗在多种癌症模型中的效果越来越被认可[16 -18 ] .在GBM和DIPG中也有很多表观转录靶向治疗策略的提出,如HDACi[19 -20 ] 、CDK7i[6 ,18 ] 、CDK9i和BETi[21 -22 ] 等.2种HGGs在发病机制和分子特性上存在许多差异,但也有很多共同的表观转录靶点.CDK12 和CDK13 (CDK12/13 )是CDK家族中主要发挥转录调控功能的2个成员,具有很高的蛋白相似性以及功能的冗余性[23 ] .它们分别与细胞周期蛋白cyclin K结合后皆可催化RNA聚合酶Ⅱ催化亚基(RNA polymerase Ⅱ subunit B1,RPB1)的C端结构域(carboxy-terminal domain,CTD)七肽重复单位中第二位丝氨酸(Ser2,S2)的磷酸化,进而通过调控转录延伸影响基因表达[24 ] .此外,它们对RNA剪接、mRNA 3′ 末端加工、内含子多聚腺苷酸化和翻译等过程也具有调控作用[23 ,25 ] .靶向抑制CDK12/13 在三阴性乳腺癌[26 ] 、肝癌[27 ] 、结直肠癌[28 ] 、胃癌[29 ] 、尤文肉瘤[30 ] 等肿瘤类型的预临床肿瘤模型汇总中均已被报道具有显著的抗肿瘤效应,但在高级别胶质瘤中尚未被报道.在CDK12/13抑制剂的抗肿瘤作用机制方面,DNA复制、基因转录、RNA剪切这些CDK12/13 参与调控的主要生物学功能均受到拮抗,并且在不同肿瘤类型中表现出了较高的一致性[26 ] .研究[31 ] 表明,cyclin K/CDK12复合物通过调控DNA损伤反应(DNA damage response,DDR)相关基因的表达来维持基因组的稳定性.CDK12/13抑制剂对DNA损伤修复的拮抗作用得到了较多关注并具有较大的临床潜在应用价值.已有研究发现,CDK12/13抑制剂处理能够在多类肿瘤中引起大量参与DNA损伤修复的关键基因的转录下调,从而诱导DNA损伤的累积[26 ] ;其主要作用机制是抑制CDK12/13可减少RNA聚合酶Ⅱ CTD S2位点的磷酸化水平,阻滞RNA聚合酶Ⅱ向基因3' 端的行进,使多外显子基因在内含子多聚腺苷酸位点处提前终止转录,产生功能受损的转录本及蛋白截短体,从而影响细胞相关功能,其中多个DDR通路基因被发现对该抑制反应尤其敏感[32 ] .进一步将CDK12/13抑制剂与能够显著诱导DNA损伤效应的放射治疗和化学治疗(放化疗)或多聚ADP核糖聚合酶抑制剂(poly ADP-ribose polymerase inhibitor,PARPi)等其他多类治疗手段进行组合治疗测试之后,发现它们之间能够产生显著的协同抗肿瘤效应或合成致死作用[26 ] ,说明CDK12/13抑制药物具有非常大的临床应用前景和商业开发价值.值得注意的是,目前已经报道过的多个CDK12/13靶向小分子,例如THZ531[33 ] 和SR-4835[26 ] ,均无法有效区分CDK12和CDK13高度相似的蛋白结构域.此外,目前还尚未有该类小分子药物进入人体临床试验. ...
... 接下来,进一步分析CDK12/13抑制剂对GBM和DIPG的体外治疗作用.如图3 A所示,THZ531[33 ] 和SR-4835[26 ] 这2个高选择性的CDK12/13抑制剂对DIPG细胞系DIPG17以及GBM细胞系U251、SF188和KNS42均表现出剂量依赖性的细胞活性抑制作用,IC50 均小于1 µmol/L,与已报道的其他类型肿瘤细胞的结果相似[26 -27 ,35 ] .流式细胞术检测靶向抑制CDK12/13对DIPG和GBM细胞增殖和凋亡的影响,结果显示0.1 µmol/L或1 µmol/L的SR-4835均可以显著抑制DIPG17、SF188和U251细胞增殖和促进其细胞凋亡(图3 B~D). ...
... 筛选结果显示在本研究所测试的2个DIPG和2个GBM细胞系中均有显著抑制作用的表观转录靶向小分子共17个,多为HDAC和CDK的抑制剂,并且其中大部分在DIPG或GBM中已经有过报道[6 ,19 -22 ] ,证明了筛选的可信度高.在CDK抑制剂中,本研究重点关注到了CDK12/13靶向小分子THZ531[33 ] 尚未在DIPG和GBM中被报道过.而在针对DIPG和GBM对CDK12/13 生长依赖性的评估中,DepMap数据库中GBM细胞系的依赖性分析数据以及利用DIPG_BJ02细胞系进行的表观转录靶向的CRISPR-Cas9功能基因组筛选结果,均表明GBM和DIPG细胞对CDK12/13 有显著依赖性(图1 ).在此基础上,进一步利用多个GBM和DIPG细胞系验证了无论是用基于CRISPR-Cas9的遗传学方法或是2个不同的靶向CDK12/13的小分子抑制剂(THZ531和SR-4835)均能产生显著的体外肿瘤抑制效应(图2 和图3 ).以上结果均表明CDK12/13 是很有潜力的HGGs治疗新靶标. ...
1
... 将SR-4835处理的DIPG17或SF188的细胞样本溶于TRIzol,送上海美吉生物医药科技有限公司进行RNA-seq转录组测序,对返还的原始测序结果Rawdata进行分析.首先利用trim galore工具进行原始数据的接头(adapter)类型检测和去接头,然后利用STAR工具将序列比对至参考基因组GRCh38,再利用htseq-count工具获得每个转录本的读数(counts);利用Gfold进行差异表达分析[34 ] ,获得每个基因的Gfold值,设置相应的阈值以筛选表达显著上调或下调的基因.本研究设置Gfold<-1为基因表达下调,Gfold>1为基因表达上调. ...
3
... 接下来,进一步分析CDK12/13抑制剂对GBM和DIPG的体外治疗作用.如图3 A所示,THZ531[33 ] 和SR-4835[26 ] 这2个高选择性的CDK12/13抑制剂对DIPG细胞系DIPG17以及GBM细胞系U251、SF188和KNS42均表现出剂量依赖性的细胞活性抑制作用,IC50 均小于1 µmol/L,与已报道的其他类型肿瘤细胞的结果相似[26 -27 ,35 ] .流式细胞术检测靶向抑制CDK12/13对DIPG和GBM细胞增殖和凋亡的影响,结果显示0.1 µmol/L或1 µmol/L的SR-4835均可以显著抑制DIPG17、SF188和U251细胞增殖和促进其细胞凋亡(图3 B~D). ...
... 根据药物剂量杀伤曲线结果(图3 A),选择0.1 µmol/L和1 µmol/L的SR-4835分别处理DIPG17细胞或SF188细胞20 h后的样本进行RNA-seq转录组测序,并重点对4个样本中相较于DMSO处理组表达水平显著下调的基因进行比较分析.如图4 A所示,与其他3个样本相比,0.1 µmol/L SR-4835处理的SF188细胞中表达水平显著下调的基因数量偏低,这与其细胞活性杀伤水平偏低相符合.因此,后续分析主要选择其他3个样本中表达水平显著下调基因的交集部分共994个基因作为研究对象.GO分析结果表明,这些基因显著富集的生物学过程与已知的CDK12/13参与调控的基因转录和DDR相关(图4 B).GSVA分析结果也显示,SR-4835能够显著下调DIPG17和SF188细胞中这2类生物学过程相关基因集合的转录水平,说明CDK12/13抑制剂作用于DIPG和GBM细胞时具有在靶(on-target)效应.已有研究[26 -27 ,35 ] 表明,在多个肿瘤类型中CDK12/13抑制剂能够通过显著拮抗DDR与放化疗以及其他靶向治疗手段之间产生显著的协同治疗效果.因此,对DIPG和GBM中靶向抑制CDK12/13是否能引起DDR相关基因的转录下调以及DNA损伤的积累进行进一步验证.如图4 D和5 A所示,通过RT-qPCR验证了SR-4835和THZ531能够在多个DIPG和GBM细胞系中引起系列关键DDR相关基因(TP53 、CCNK 、ATM 、ATR 、E2F1 、BRCA1 、FANCD2 、SMARCC2 、RAD51 、FANCI )的转录下调.此外,通过蛋白质印迹法(Western blotting)验证了SR-4835和THZ531处理能够显著下调DIPG和GBM细胞中RNA Pol Ⅱ CTD S2位点的磷酸化(RNA Pol Ⅱ S2P)水平,并且显著上调DNA双链断裂(double-strand break,DSB)标志物γH2AX(组蛋白H2AX第139位丝氨酸的磷酸化[36 ] )的水平,表明在这2类HGGs中靶向抑制CDK12/13确实能够引起DNA损伤的积累(图5 B). ...
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
1
... 根据药物剂量杀伤曲线结果(图3 A),选择0.1 µmol/L和1 µmol/L的SR-4835分别处理DIPG17细胞或SF188细胞20 h后的样本进行RNA-seq转录组测序,并重点对4个样本中相较于DMSO处理组表达水平显著下调的基因进行比较分析.如图4 A所示,与其他3个样本相比,0.1 µmol/L SR-4835处理的SF188细胞中表达水平显著下调的基因数量偏低,这与其细胞活性杀伤水平偏低相符合.因此,后续分析主要选择其他3个样本中表达水平显著下调基因的交集部分共994个基因作为研究对象.GO分析结果表明,这些基因显著富集的生物学过程与已知的CDK12/13参与调控的基因转录和DDR相关(图4 B).GSVA分析结果也显示,SR-4835能够显著下调DIPG17和SF188细胞中这2类生物学过程相关基因集合的转录水平,说明CDK12/13抑制剂作用于DIPG和GBM细胞时具有在靶(on-target)效应.已有研究[26 -27 ,35 ] 表明,在多个肿瘤类型中CDK12/13抑制剂能够通过显著拮抗DDR与放化疗以及其他靶向治疗手段之间产生显著的协同治疗效果.因此,对DIPG和GBM中靶向抑制CDK12/13是否能引起DDR相关基因的转录下调以及DNA损伤的积累进行进一步验证.如图4 D和5 A所示,通过RT-qPCR验证了SR-4835和THZ531能够在多个DIPG和GBM细胞系中引起系列关键DDR相关基因(TP53 、CCNK 、ATM 、ATR 、E2F1 、BRCA1 、FANCD2 、SMARCC2 、RAD51 、FANCI )的转录下调.此外,通过蛋白质印迹法(Western blotting)验证了SR-4835和THZ531处理能够显著下调DIPG和GBM细胞中RNA Pol Ⅱ CTD S2位点的磷酸化(RNA Pol Ⅱ S2P)水平,并且显著上调DNA双链断裂(double-strand break,DSB)标志物γH2AX(组蛋白H2AX第139位丝氨酸的磷酸化[36 ] )的水平,表明在这2类HGGs中靶向抑制CDK12/13确实能够引起DNA损伤的积累(图5 B). ...
2
... 如图6 A、B所示,RNA-seq转录组测序数据的GO和GSVA分析还发现SR-4835分别处理DIPG17和SF188细胞之后均能够显著下调蛋白酶体-泛素化与细胞周期相关的生物学功能.已有研究[37 ] 表明DNA损伤的累积与泛素-蛋白酶体功能的下调均可以诱导肿瘤细胞阻滞在G2-M期并发生凋亡,因此,接下来用0.1 µmol/L和1 µmol/L的SR-4835分别处 ...
... 为了探究HGGs中靶向抑制CDK12/13 拮抗肿瘤的分子机制,对SR-4835处理的DIPG细胞系DIPG17和pGBM细胞系SF188的RNA-seq结果进行分析,重点关注在2个细胞系中均表达下调的基因(Gfold<-1).GO分析表明,这些基因显著富集于细胞周期、基因转录、RNA剪切以及DNA修复等已知的CDK12/13参与调控的多个生物学过程或功能;进一步的GSVA分析也证明了以上生物学过程或功能受到CDK12/13抑制剂的拮抗;再结合在SR-4835和THZ531处理的多个GBM和DIPG细胞系中所检测到的RNA Pol Ⅱ S2P水平下降,均说明研究所使用的CDK12/13靶向小分子抑制剂发挥了on-target的抑制作用.在此基础上,本研究首先针对CDK12/13抑制剂引起的DNA损伤修复功能失调进行了验证.已有研究[26 ] 发现肿瘤细胞中的多个DNA损伤修复基因对CDK12/13抑制剂引起的转录下调非常敏感,这与本研究在多个HGGs细胞系中的RT-qPCR验证结果一致(图5 A).同时,检测到了双链DNA损伤标志物γH2AX蛋白磷酸化水平在CDK12/13抑制剂处理之后发生上调(图5 B),说明确实发生了DNA损伤的累积.GSVA分析结果还表明CDK12/13抑制剂主要下调双链DDR和同源重组修复相关基因(图4 C).以上结果均提示在GBM和DIPG中,CDK12/13抑制剂可能也像以往在众多肿瘤模型中报道[38 -39 ] 过的那样,能够与放化疗或者PARPi产生协同抑制效应或合成致死效应.此外,已有研究[40 ] 表明当DNA损伤不能得到及时修复时,细胞将启动G2-M期DNA损伤检查点,防止损伤细胞进入有丝分裂,这与检测到的CDK12/13抑制剂可引起DIPG和GBM细胞发生G2-M期细胞周期阻滞的结果相符合(图6 C).同时,RNA-seq分析还发现了CDK12/13抑制剂可引起蛋白酶体介导的泛素化蛋白降解过程相关基因的表达显著下调(图6 A).而根据以往报道[37 ] ,靶向抑制蛋白酶体功能同样可以使肿瘤细胞发生G2-M期阻滞.综上所述,本研究推测抑制CDK12/13导致DDR下调和蛋白酶体介导的泛素化蛋白降解减少很可能共同构成细胞G2-M期阻滞的上游分子事件.另一方面,本研究注意到GBM和DIPG细胞受CDK12抑制剂处理后各自存在很多特有的表达显著下调基因,这提示不同的HGGs细胞可能对CDK12抑制剂具有一些差异性的反应(图4 A).因此,进一步挖掘不同肿瘤类型特有的CDK12靶向效应及相关分子机制将有望为该类药物的临床应用提供更精准的指导和参考. ...
1
... 为了探究HGGs中靶向抑制CDK12/13 拮抗肿瘤的分子机制,对SR-4835处理的DIPG细胞系DIPG17和pGBM细胞系SF188的RNA-seq结果进行分析,重点关注在2个细胞系中均表达下调的基因(Gfold<-1).GO分析表明,这些基因显著富集于细胞周期、基因转录、RNA剪切以及DNA修复等已知的CDK12/13参与调控的多个生物学过程或功能;进一步的GSVA分析也证明了以上生物学过程或功能受到CDK12/13抑制剂的拮抗;再结合在SR-4835和THZ531处理的多个GBM和DIPG细胞系中所检测到的RNA Pol Ⅱ S2P水平下降,均说明研究所使用的CDK12/13靶向小分子抑制剂发挥了on-target的抑制作用.在此基础上,本研究首先针对CDK12/13抑制剂引起的DNA损伤修复功能失调进行了验证.已有研究[26 ] 发现肿瘤细胞中的多个DNA损伤修复基因对CDK12/13抑制剂引起的转录下调非常敏感,这与本研究在多个HGGs细胞系中的RT-qPCR验证结果一致(图5 A).同时,检测到了双链DNA损伤标志物γH2AX蛋白磷酸化水平在CDK12/13抑制剂处理之后发生上调(图5 B),说明确实发生了DNA损伤的累积.GSVA分析结果还表明CDK12/13抑制剂主要下调双链DDR和同源重组修复相关基因(图4 C).以上结果均提示在GBM和DIPG中,CDK12/13抑制剂可能也像以往在众多肿瘤模型中报道[38 -39 ] 过的那样,能够与放化疗或者PARPi产生协同抑制效应或合成致死效应.此外,已有研究[40 ] 表明当DNA损伤不能得到及时修复时,细胞将启动G2-M期DNA损伤检查点,防止损伤细胞进入有丝分裂,这与检测到的CDK12/13抑制剂可引起DIPG和GBM细胞发生G2-M期细胞周期阻滞的结果相符合(图6 C).同时,RNA-seq分析还发现了CDK12/13抑制剂可引起蛋白酶体介导的泛素化蛋白降解过程相关基因的表达显著下调(图6 A).而根据以往报道[37 ] ,靶向抑制蛋白酶体功能同样可以使肿瘤细胞发生G2-M期阻滞.综上所述,本研究推测抑制CDK12/13导致DDR下调和蛋白酶体介导的泛素化蛋白降解减少很可能共同构成细胞G2-M期阻滞的上游分子事件.另一方面,本研究注意到GBM和DIPG细胞受CDK12抑制剂处理后各自存在很多特有的表达显著下调基因,这提示不同的HGGs细胞可能对CDK12抑制剂具有一些差异性的反应(图4 A).因此,进一步挖掘不同肿瘤类型特有的CDK12靶向效应及相关分子机制将有望为该类药物的临床应用提供更精准的指导和参考. ...
1
... 为了探究HGGs中靶向抑制CDK12/13 拮抗肿瘤的分子机制,对SR-4835处理的DIPG细胞系DIPG17和pGBM细胞系SF188的RNA-seq结果进行分析,重点关注在2个细胞系中均表达下调的基因(Gfold<-1).GO分析表明,这些基因显著富集于细胞周期、基因转录、RNA剪切以及DNA修复等已知的CDK12/13参与调控的多个生物学过程或功能;进一步的GSVA分析也证明了以上生物学过程或功能受到CDK12/13抑制剂的拮抗;再结合在SR-4835和THZ531处理的多个GBM和DIPG细胞系中所检测到的RNA Pol Ⅱ S2P水平下降,均说明研究所使用的CDK12/13靶向小分子抑制剂发挥了on-target的抑制作用.在此基础上,本研究首先针对CDK12/13抑制剂引起的DNA损伤修复功能失调进行了验证.已有研究[26 ] 发现肿瘤细胞中的多个DNA损伤修复基因对CDK12/13抑制剂引起的转录下调非常敏感,这与本研究在多个HGGs细胞系中的RT-qPCR验证结果一致(图5 A).同时,检测到了双链DNA损伤标志物γH2AX蛋白磷酸化水平在CDK12/13抑制剂处理之后发生上调(图5 B),说明确实发生了DNA损伤的累积.GSVA分析结果还表明CDK12/13抑制剂主要下调双链DDR和同源重组修复相关基因(图4 C).以上结果均提示在GBM和DIPG中,CDK12/13抑制剂可能也像以往在众多肿瘤模型中报道[38 -39 ] 过的那样,能够与放化疗或者PARPi产生协同抑制效应或合成致死效应.此外,已有研究[40 ] 表明当DNA损伤不能得到及时修复时,细胞将启动G2-M期DNA损伤检查点,防止损伤细胞进入有丝分裂,这与检测到的CDK12/13抑制剂可引起DIPG和GBM细胞发生G2-M期细胞周期阻滞的结果相符合(图6 C).同时,RNA-seq分析还发现了CDK12/13抑制剂可引起蛋白酶体介导的泛素化蛋白降解过程相关基因的表达显著下调(图6 A).而根据以往报道[37 ] ,靶向抑制蛋白酶体功能同样可以使肿瘤细胞发生G2-M期阻滞.综上所述,本研究推测抑制CDK12/13导致DDR下调和蛋白酶体介导的泛素化蛋白降解减少很可能共同构成细胞G2-M期阻滞的上游分子事件.另一方面,本研究注意到GBM和DIPG细胞受CDK12抑制剂处理后各自存在很多特有的表达显著下调基因,这提示不同的HGGs细胞可能对CDK12抑制剂具有一些差异性的反应(图4 A).因此,进一步挖掘不同肿瘤类型特有的CDK12靶向效应及相关分子机制将有望为该类药物的临床应用提供更精准的指导和参考. ...
1
... 为了探究HGGs中靶向抑制CDK12/13 拮抗肿瘤的分子机制,对SR-4835处理的DIPG细胞系DIPG17和pGBM细胞系SF188的RNA-seq结果进行分析,重点关注在2个细胞系中均表达下调的基因(Gfold<-1).GO分析表明,这些基因显著富集于细胞周期、基因转录、RNA剪切以及DNA修复等已知的CDK12/13参与调控的多个生物学过程或功能;进一步的GSVA分析也证明了以上生物学过程或功能受到CDK12/13抑制剂的拮抗;再结合在SR-4835和THZ531处理的多个GBM和DIPG细胞系中所检测到的RNA Pol Ⅱ S2P水平下降,均说明研究所使用的CDK12/13靶向小分子抑制剂发挥了on-target的抑制作用.在此基础上,本研究首先针对CDK12/13抑制剂引起的DNA损伤修复功能失调进行了验证.已有研究[26 ] 发现肿瘤细胞中的多个DNA损伤修复基因对CDK12/13抑制剂引起的转录下调非常敏感,这与本研究在多个HGGs细胞系中的RT-qPCR验证结果一致(图5 A).同时,检测到了双链DNA损伤标志物γH2AX蛋白磷酸化水平在CDK12/13抑制剂处理之后发生上调(图5 B),说明确实发生了DNA损伤的累积.GSVA分析结果还表明CDK12/13抑制剂主要下调双链DDR和同源重组修复相关基因(图4 C).以上结果均提示在GBM和DIPG中,CDK12/13抑制剂可能也像以往在众多肿瘤模型中报道[38 -39 ] 过的那样,能够与放化疗或者PARPi产生协同抑制效应或合成致死效应.此外,已有研究[40 ] 表明当DNA损伤不能得到及时修复时,细胞将启动G2-M期DNA损伤检查点,防止损伤细胞进入有丝分裂,这与检测到的CDK12/13抑制剂可引起DIPG和GBM细胞发生G2-M期细胞周期阻滞的结果相符合(图6 C).同时,RNA-seq分析还发现了CDK12/13抑制剂可引起蛋白酶体介导的泛素化蛋白降解过程相关基因的表达显著下调(图6 A).而根据以往报道[37 ] ,靶向抑制蛋白酶体功能同样可以使肿瘤细胞发生G2-M期阻滞.综上所述,本研究推测抑制CDK12/13导致DDR下调和蛋白酶体介导的泛素化蛋白降解减少很可能共同构成细胞G2-M期阻滞的上游分子事件.另一方面,本研究注意到GBM和DIPG细胞受CDK12抑制剂处理后各自存在很多特有的表达显著下调基因,这提示不同的HGGs细胞可能对CDK12抑制剂具有一些差异性的反应(图4 A).因此,进一步挖掘不同肿瘤类型特有的CDK12靶向效应及相关分子机制将有望为该类药物的临床应用提供更精准的指导和参考. ...
2
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
... [41 ].通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
1
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
1
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
1
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...
1
... 已有研究[27 ,29 ,35 ] 报道在多个肿瘤预临床模型的体内治疗中,CDK12/13抑制剂确实通过下调DDR和诱导细胞周期阻滞发挥抑制作用,且可以与化疗药或者PARPi之间存在协同抗肿瘤作用.目前本研究相关的功能验证和靶向治疗均是基于细胞模型的体外实验,在今后的工作中本研究将进一步通过相应的体内实验来验证靶向抑制CDK12/13针对GBM和DIPG的治疗潜力,尤其是与现有放化疗之间的组合效应.值得注意的是,血脑屏障的存在限制了很多小分子药物进入脑部区域[41 ] ,使得GBM和DIPG的药物治疗面临更多的挑战.现有的CDK12/13抑制剂THZ531和SR-4835均未报道能通过血脑屏障.因此,未来CDK12/13抑制剂药物的临床应用还需要克服血脑屏障的问题.要解决这个问题,可开发新的可以通过血脑屏障的靶向小分子,或者依赖药物递送系统实现颅内给药.载药纳米颗粒对于中枢神经系统疾病的治疗已经过美国FDA批准进入临床试验阶段[41 ] .通过对流增强递送(convection enhanced delivery,CED)可将载药纳米颗粒注入局部肿瘤实现颅内给药[42 ] ;细胞穿透肽(cell penetrating peptide,CPP)的阳离子电荷通过静电相互作用与脑内皮细胞膜表面结合有助于CPP修饰的纳米材料通过血脑屏障,将药物运输到大脑[43 ] .载药纳米颗粒传递系统在DIPG和GBM中均有报道[44 -45 ] ,因此该方法有望大大促进CDK12/13靶向新策略的体内验证与临床转化. ...








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}