

哺乳动物早期发育过程受到一系列复杂且精密的调控。小鼠和人类的生命均始于卵子受精。在小鼠胚胎3.5 d时,合子通过细胞增殖和分化产生滋养外胚层和内细胞团。内细胞团进而分化成上胚层和原始内胚层。上胚层前部发育成外胚层(ectoderm),而其后部则形成原条。原条是向中胚层(mesoderm)和内胚层(endoderm)分化的中间过渡阶段,因此又被称为中内胚层(mesendoderm)阶段。
小鼠胚胎干细胞(mouse embryonic stem cell,mESC)来源于内细胞团[1]。多项研究发现白血病抑制因子(leukemia inhibitory factor,LIF)是稳定维持和培养mESC最重要的蛋白分子[2]。在mESC中约有0.5%的细胞类似于胚胎二细胞阶段的二细胞样细胞(2-cell-like cell,2CLC),表达双向同源框因子(double homeobox,Dux)、锌指和扫描结构域4因子(zinc finger and scan domain containing 4,Zscan4)以及Zscan4d等[3]。当mESC在无LIF的悬浮培养基中,需要先形成类胚体(embryoid body,EB),再逐步分化为3个胚层[4]。EB分化的过程在体外重现了体内的胚层分化。因此胚胎干细胞是研究早期胚胎细胞分化命运选择机制的经典模型。
胚胎干细胞具有多能性和自我更新的特点,其干性维持主要由多能性因子和自我更新信号通路等共同调控[5]。但随着多能性因子与分化调控因子间平衡被打破,干细胞的命运便发生改变,多能性逐渐丢失,进一步分化成不同谱系的细胞。当mESC退出多能性状态1~2 d后,关键中内胚层分化因子T-box转录因子Brachyury(即T基因)等率先开始上调,之后其他分化基因如GSC同源盒(goosecoid homeobox,Gsc)才会逐渐被诱导表达[6],从而正式启动中内胚层分化。因此,解析T基因的调控网络将有助于阐明干细胞向中内胚层分化的关键机制。如果能在mESC中构建T基因的增强绿色荧光蛋白(enhanced green fluorescent protein,EGFP)报告系统,那么当mESC形成EB进入三胚层分化的情况下,T报告基因的荧光信号就会随着T基因的表达增强而逐渐显露,从而实时反映中内胚层分化情况。
目前,CRISPR/Cas9(clustered regularly interspaced short palindromic repeat/CRISPR associated protein 9)技术已成为研究干细胞功能的重要手段[7]。利用CRISPR/Cas9和同源定向修复(homologous-directed repair,HDR)技术,可以在特定靶基因位点敲入转基因[8],构建携带特定转基因的细胞系。但该技术仍存在频繁脱靶现象,于是CRISPR/Cas9n(Cas9 nickase,Cas9n)应运而生。CRISPR/Cas9n系统能诱导DNA发生单链切口而不是双链断裂。通常在真核细胞中,单链缺口会被高保真修复,因此其脱靶率被大大降低[9]。此外,当需要在细胞中同时表达2个或以上的基因时,2A肽段[例如手足口病毒2A(foot-and-mouth disease virus 2A,F2A)[10]]常常被置于需要同时表达的基因之间。当核糖体从上游基因到达F2A序列的C末端时,跳过了甘氨酸-脯氨酰肽键的合成。这种核糖体跳跃导致了F2A和下游肽的断裂,从而保证F2A上下游基因在不产生融合的情况下又能同时独立表达[11]。
本研究旨在利用CRISPR/Cas9n技术在mESC的T基因末端依次敲入F2A和EGFP,用以建立T荧光报告细胞系(mT-F2A-EGFP)。EGFP的荧光强度可实时反映中内胚层细胞分化情况,这将为深入研究中内胚层早期分化调控机制和互作网络奠定基础。
1 材料与方法
1.1 材料
1.1.1 细胞
mEST系E14Tg2a(E14细胞)购自美国ATCC公司。
1.1.2 质粒
pX461-Cas9n-2A-GFP(#48140)购自美国Addgene公司。
1.1.3 主要试剂
菌株Trans10化学感受态细胞(#CD201)和反转录试剂盒(#AT341-02)购自中国全式金公司;Knockout DMEM(Dulbecco's Modified Eagle Medium)培养基(10829-018)、DMEM+GlutaMAX培养基(10569-010)、丙酮酸钠(11360-070)、55 mmol/L 2-巯基乙醇(21985023)、TrypLE(12605028)、胎牛血清(fetal bovine serum,FBS;5H30070.03)、青霉素/链霉素(15140-122)均购自美国Thermo Fisher Scientific公司;小鼠LIF(#ESG1107)购自美国Millipore公司;0.1%明胶(#07903)购自加拿大Stem Cell公司;小鼠胚胎干细胞核转染试剂盒(#VPH-1001)购自瑞士Lonza公司;Hieff Canace高保真DNA聚合酶(10148ES60)购自中国翊圣公司;T4 DNA连接酶(B0202S)和Gibson组装预混液(E2611)购自美国NEB公司;去内毒素质粒大量提取试剂盒(740420.50)、NucleoZol RNA提取试剂(740404.200)购自德国MN公司;PI染色液(#P4171)购自北京Lablead公司;碱性磷酸酶染色试剂盒(Vector Blue Substrate Kit,Alkaline Phosphatase;#SK-5300)购自美国Vector Laboratories公司。
1.2 方法
1.2.1 质粒构建
(1) 构建pX461-Cas9n-2A-Hygro质粒
利用Fse1和Sbf1的酶切位点移除pX461-Cas9n-2A-GFP质粒的GFP片段,并利用Gibson试剂插入抗潮霉素DNA片段。Gibson组装方法:将0.5 pmol DNA片段和10 μL Gibson Assembly Master Mix混合,加入双蒸水(ddH2O)补齐20 μL,在50 °C放置1 h。
(2) 构建pX461-mT-sgRNA质粒
挑选小鼠T基因终止密码子附近的靶序列(GTGCTGAGACTTG-TAACAAC)设计单链导向RNA(single guide RNA,sgRNA)并通过互补链退火形成mT-sgRNA双链,再插入pX461-Cas9n-2A-Hygro质粒中。反应体系:0.2 μL 100 ng pX461-Cas9n-2A-Hygro,2 μL mT-sgRNA双链(1∶200稀释),2 μL Tango缓冲液,1 μL 10 mmol/L DTT,1 μL 10 mmol/L ATP,1 μL FastDigest Bbs1,0.5 μL T4 DNA连接酶,加入ddH2O补齐至20 μL。在PCR仪器上进行反应:37 ℃ 5 min,23 ℃ 5 min,重复6个循环。
(3) 构建mT-F2A-EGFP-Donor供体质粒
利用PCR技术分别扩增出EGFP和杀稻瘟菌素-S脱氨酶(blasticidin-S deaminas,BSD),并且将小鼠基因组中T基因的终止密码子上下游各1 000 bp分别扩增,得到左同源臂(left homologous arm,HAL)和右同源臂(right homologous arm,HAR)片段。通过Gibson试剂把这些片段组装相连。
1.2.2 细胞培养与单克隆筛选
提前在培养皿中加入0.1%明胶,10 min后去除。然后将所需的E14细胞和3 mL培养基混合后静置于37 °C二氧化碳培养箱中培养。E14培养基成分:Knockout DMEM、丙酮酸钠(100 mmol/L)、MEM-NEAA、胎牛血清、青霉素/链霉素溶液、两性霉素B、谷氨酰胺(200 mmol/L)、β-巯基乙醇(55 mmol/L)和LIF(107 unit/mL)。EB分化实验的培养基中不添加LIF,将6×105个E14细胞种于低吸附培养皿中,持续培养4 d。每2 d更换1次培养液。
将pX461-mT-sgRNA重组质粒和mT-F2A-EGFP-Donor供体质粒按4∶6的比例,使用小鼠胚胎干细胞核转染试剂盒,对2×106个细胞进行电转。电转24~48 h后,用带有潮霉素(hygromycin,Hygro)和BSD的含药培养基进行药筛培养5 d。将药筛后存活的细胞按照每孔1个细胞种入96孔板中,在培养约10 d后,即可收取单克隆细胞系。采用PCR进行鉴定,测序引物序列为T-HAL-F(GATTGAGCAGTAG-TGGTCTA)、T-EGFP-R(CGTCCTTGAAGAAGATG-GT)、T-BSD-F(CCACATACACTTCATTCTCAG)、T-HAR-R(TCAGCCTCTTCCTATCTCAT)、mT-F(CT-GGTCTGTGAGCAATGG)、mT-R(CTCTGTCCTT-GGCTTCATAA)。
1.2.3 RNA的提取
利用500 μL NucleoZOL充分裂解细胞,随后加入200 μL ddH2O,上下剧烈振荡15 s,12 000×g离心15 min后,将含有RNA的上清液转移至干净的1.5 mL EP管中,加入等体积异丙醇沉淀RNA,上下颠倒混匀后,室温静置10 min,12 000×g离心10 min。去上清液后,分别用75%和100%乙醇清洗1次,晾干后加入干净的DEPC水溶解RNA,随后定量。使用反转录试剂盒将RNA反转录为cDNA。
1.2.4 实时定量聚合酶链反应
按如下反应体系配制实时定量聚合酶链反应(real-time quantitative reverse transcription polymerase chain reaction,RT-qPCR)的混合液:5 μL PowerUp™ SYBR™ Green预混液、2.5 μL 1∶10稀释后的cDNA、1 μL 1 μmol/L上下游引物和1.5 μL ddH2O,在Bio-rad荧光定量PCR仪上检测。RT-qPCR的引物序列见表1。
表1 RT-qPCR引物序列
Tab 1
| Gene | Forward primer (5'→3') | Reverse primer (5'→3') |
|---|---|---|
| Gapdh | CTCCACTCACGGCAAATTCA | CGCTCCTGGAAGATGGTGAT |
| Sox2 | GCGGAGTGGAAACTTTTGTCC | CGGGAAGCGTGTACTTATCCTT |
| Gsc | TTGCACAGACAGTCGATGCTACT | TCGTTGCTTTCTCGACCCC |
| T | TCCTCCATGTGCTGAGACTTGT | CCAAGAGCCTGCCACTTTG |
| Eomes | GCGCATGTTTCCTTTCTTGAG | GGTCGGCCAGAACCACTTC |
| nestin | CAGGATTGGGAGGAGGGCAGAG | GGAGGCAGGAGACTTCAGGTAG |
| Pax6 | GTTCCCTGTCCTGTGGACTC | ACCGCCCTTGGTTAAAGTCT |
| Sox1 | ATACCGCAATCCCCTCTCAG | ACAACATCCGACTCCTCTTCC |
| Dux | CCCAGCGACTCAAACTCCTTC | GGACTTCGTCCAGCAGTTGAT |
| Zscan4 | GAGATTCATGGAGAGTCTGACTGATGAGTG | GCTGTTGTTTCAAAAGCTTGATGACTTC |
| Zscan4d | GTCCTGACAGAGGCCTGCC | GAGATGTCTGAAGAGGCAAT |
1.2.5 荧光显微镜的使用
使用尼康倒置荧光显微成像系统,将培养皿平放在载物台上。10倍物镜下选择合适的滤光片,对样品的荧光表达强度进行观察和拍照记录。
1.2.6 碱性磷酸酶染色
对6孔板中的细胞进行固定和碱性磷酸酶(alkaline phosphatase,AP)染色。每孔加入1 mL 4%多聚甲醛固定,室温静置10 min。配制染色液:在5 mL pH为8.2~8.5的100 mmol/L Tris-HCl溶液中加入2滴Vector Blue Reagent 1、2滴Vector Blue Reagent 2和2滴Vector Blue Reagent 3,充分混匀后使用。将6孔板中的4%多聚甲醛吸干,加入预冷的PBS洗2次后,加入预先配好的染色液,在显微镜下观察到蓝色即可终止染色并拍照。
1.2.7 细胞周期检测
收取2×106个细胞于15 mL离心管中,加入预冷的PBS洗2次。使用预冷的70%乙醇4 °C固定过夜。然后,4 ℃ 160×g离心5 min。PBS洗2次后加入终浓度为100 μg/mL的RNase A,于37 ℃反应30 min。360×g离心5 min,弃上清液,加入终浓度为50 μg/mL PI染料,室温避光15 min后,上流式细胞仪进行细胞周期检测。然后使用FlowJo Version 10.8软件和Modfit LT Version 5.0软件对流式数据进行处理。
1.2.8 生长曲线
在24孔板每孔接种5×103个细胞,进行常规细胞培养6 d。每24 h换液1次,并取样、计数。每次取3个孔的细胞,多次计数取平均值以提高数据准确性。以天数为横坐标,以细胞数为纵坐标,绘制散点图,得到细胞生长曲线。
1.3 统计学分析
通过GraphPad Prism 9.0软件进行统计学分析。所有实验均进行3次以上独立重复实验。数据以x±s的形式表示。组间比较采用独立样本t检验。P<0.05表示差异具有统计学意义。
2 结果
2.1 构建pX461-mT-sgRNA和mT-F2A-EGFP-Donor质粒
图1
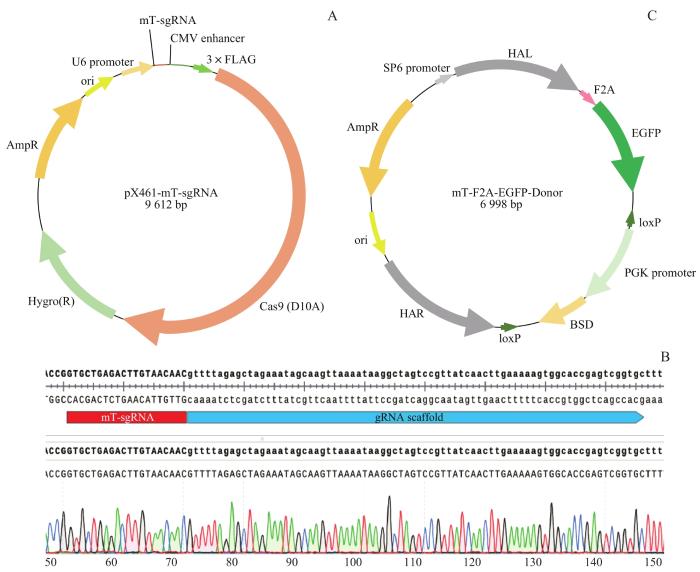
图1
pX461-mT-sgRNA质粒和mT-F2A-EGFP-Donor质粒的构建
Note:A. Scheme of pX461-mT-sgRNA plasmid map. B. Sequencing result of mT-sgRNA cloned into pX461-Cas9n-2A-Hygro plasmid. C. Scheme of mT-F2A-EGFP-Donor plasmid map.
Fig 1
Construction of pX461-mT-sgRNA plasmid and mT-F2A-EGFP-Donor plasmid
mT-F2A-EGFP-Donor质粒(图1C)可以为DNA修复提供模板(template),从而在T基因终止密码子之前插入F2A-EGFP标记。T终止密码子前后约1 000 bp的HAL和HAR分别连接在F2A-EGFP的上下游。而PGK启动子驱动的BSD药物筛选标记可以提高重组单克隆细胞的筛选效率。此外,在PGK-BSD的两侧各有一个同方向的loxP(locus of X-overP1)序列。在挑选到正确的mT-F2A-EGFP克隆后,可以通过重组酶系统(cyclization recombination enzyme,Cre)使2个loxP位点间发生基因重组,从而有效删除2个loxP位点间的PGK-BSD序列。
2.2 共转染pX461-mT-sgRNA和mT-F2A-EGFP-Donor质粒
通过电穿孔将pX461-mT-sgRNA和mT-F2A-EGFP-Donor质粒瞬转到E14细胞中。mT-sgRNA引导Cas9n蛋白在T基因的终止密码子附近进行了单链切割,所产生的单链缺口则以mT-F2A-EGFP-Donor质粒为模板进行高保真修复,从而在基因组上T基因终止密码子之前插入F2A-EGFP和BSD标记。当T基因被转录表达时,F2A序列确保了EGFP和BSD的同时表达(图2A)。电转后24 h,转染组细胞在形态上与对照组有所不同,部分细胞呈现出扁平或者梭形的应激状态。而对照组细胞保持克隆样隆起的集落。值得注意的是,在添加了Hygro和杀稻瘟菌素的培养基中培养3 d时,稳转成功的细胞由于存在抗药性而保持正常生长,而对照组细胞大部分死亡(图2B)。这就证明F2A-EGFP-BSD序列已经通过HDR的方式整合进入了T基因序列中。
图2
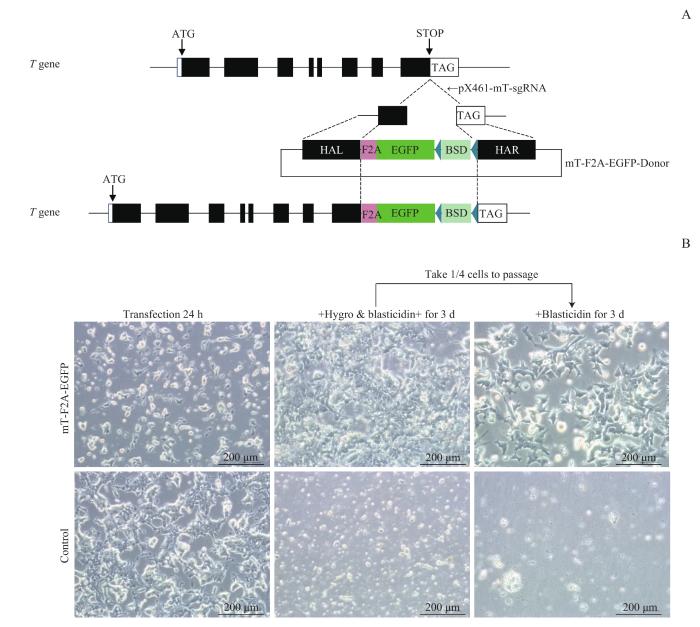
图2
共转染pX461-mT-sgRNA和mT-F2A-EGFP-Donor质粒
Note: A. The scheme showed that F2A-EGFP was inserted before the stop codon of T gene. B. The cell morphology of E14 cells 24 h after electroporation with 2 kinds of plasmids (upper left), 3 d after Hygro and blasticidin selection (middle upper) and another 3 d with blasticidin (upper right). The untransfected E14 cells were used as a control (bottom).
Fig 2
Co-transfection of pX461-mT-sgRNA and mT-F2A-EGFP-Donor plasmid
2.3 mT-F2A-EGFP单克隆细胞的鉴定
图3
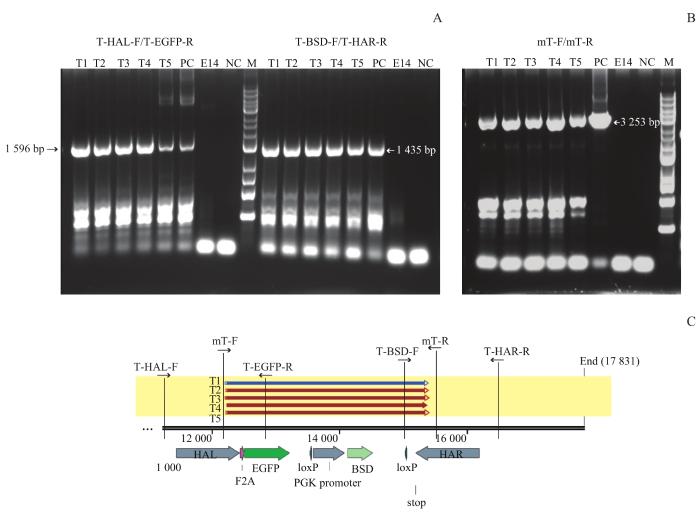
图3
mT-F2A-EGFP单克隆细胞的鉴定
Note:A. Gel electrophoresis of PCR products with primers of T-HAL-F/T-EGFP-R (left, 1 596 bp ) or T-BSD-F/T-HAL-R (right, 1 435 bp). B. Gel electrophoresis of PCR products with primers of mT-F/mT-R (3 253 bp). C. Sequencing results of T1, T2, T3, T4 and T5 clones. Primers used for PCR were also labeled here. PC—positive control; NC—negative control; M—marker.
Fig 3
Identification of mT-F2A-EGFP clones
2.4 mT-F2A-EGFP荧光报告干细胞系具有正常的分化功能
通过上述实验已经获得了5个候选单克隆细胞系。接下来进行EB分化实验来鉴定这些单克隆细胞的多能性和分化功能是否正常。在分化过程中,T基因作为mESC向中内胚层发育的关键调控因子,其表达量会随着EB分化的进行而逐渐升高。野生型E14和mT-F2A-EGFP报告细胞系T1~T5均能在不含mLIF的悬浮培养基中逐渐形成EB,且形态上并无明显异常。收取未分化的干细胞RNA(D0),以及EB分化后第3日(D3)和第4日(D4)的RNA,进行RT-qPCR实验检测多能性标志基因以及各胚层标志基因的表达情况,结果显示除了T2和T3的分化能力异常之外,其余3个候选单克隆细胞系T1、T4和T5的EB分化能力和野生型E14并无区别:随着分化的进行,它们在转录水平上表现出多能性基因性别决定域Y框蛋白2(Sox2)的下调,中内胚层分化基因T、脱中胚蛋白(Eomes)和Gsc,以及外胚层基因巢蛋白(nestin)的上调。其中,单克隆T1的多能性基因以及中内胚层、外胚层基因的表达量均与正常对照组E14最为接近(图4)。因此推测,在T1克隆中F2A-EGFP和T基因的重组对于E14细胞的干性维持和分化能力无明显影响。
图4
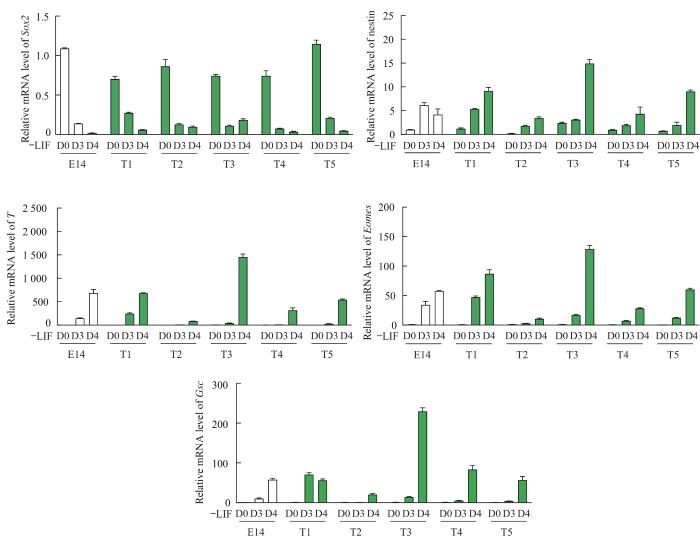
图4
mT-F2A-EGFP单克隆细胞分化过程中各标记基因的相对表达量
Note: RT-qPCR experiments show relative mRNA levels of pluripotent marker (Sox2), mesendoderm marker (T, Eomes and Gsc), and ectoderm marker (nestin). RNA of E14, T1, T2, T3, T4 and T5 clones were collected before differentiation (D0), and on the 3rd (D3) and the 4th (D4) day of EB differentiation.
Fig 4
Relative expression levels of marker genes during lineage differentiation of mT-F2A-EGFP clones
2.5 mT-F2A-EGFP荧光报告干细胞克隆的鉴定
为验证mT-F2A-EGFP荧光报告细胞系的构建是否成功,分别从形态学、分化时T荧光表达强度、AP活性、生长曲线和细胞周期等方面进行检测。首先,挑选E14、T1、T2、T3、T4和T5克隆进行EB分化实验。在添加LIF的情况下,E14和T报告细胞系均呈边界平滑的克隆状生长;在不含LIF的培养基中,均被诱导形成大小相似的球状EB(图5A,D0)。在荧光显微镜下,野生型E14细胞由于没有T荧光报告基因,无论是分化前D0,还是分化后D3和D4均不表达T绿色荧光(图5A);而在携带mT-EGFP报告基因的克隆细胞中,只有T1、T3和T4的荧光表达强度与T基因的表达水平呈现正相关:D0时,T基因不表达,因此没有EGFP荧光被检测到;但随着T基因的表达上升,T1、T3和T4在分化后D3开始呈现绿荧光,并在D4时mT-EGFP荧光到达高峰(图5A)。这证明在分化前后mT-EGFP的荧光动态变化和T基因的表达水平高度一致(图4和图5A),尤其是T1克隆的荧光信号最强。这一现象在流式细胞分析中得到了进一步的验证。当分化到D3和D4时,EB中包含了多种谱系的细胞[4-5];以野生型E14为阴性对照,利用流式细胞技术从T1、T3和T4细胞系分化的多谱系细胞群中分选出mT-F2A-EGFP荧光表达细胞(EGFP+)和无荧光表达的细胞(EGFP-)。通过分析荧光表达细胞的比例可以更直观地证明,T1、T3和T4细胞系的EGFP+细胞在分化D3时出现,并在分化D4时显著增多。值得注意的是,克隆T1在D4时有高达90.47%的EGFP+细胞,并且该克隆的多能性基因和三胚层分化基因,包括T基因的表达水平和野生型E14最为一致(图4),因此T1报告细胞系能通过EGFP的荧光强度实时准确地反映出EB中内源T基因的表达情况(图4,图5A、5B)。同时,E14细胞以及3个克隆报告细胞系均具有较高的AP染色活性(图5C),说明F2A-EGFP的敲入对E14细胞的干性维持无明显不良影响。另外,T1、T3和T4细胞的生长曲线和E14的生长曲线并无明显区别(图5D)。并且,和E14相比,这些克隆细胞的细胞周期均分布正常(图5E)。总之,本研究基于CRISPR/Cas9n技术成功建立了携带mT-F2A-EGFP报告系统的小鼠胚胎干细胞系,其中T1克隆无论是在基因表达水平还是在荧光强度上都能准确反映T基因的表达情况。
图5
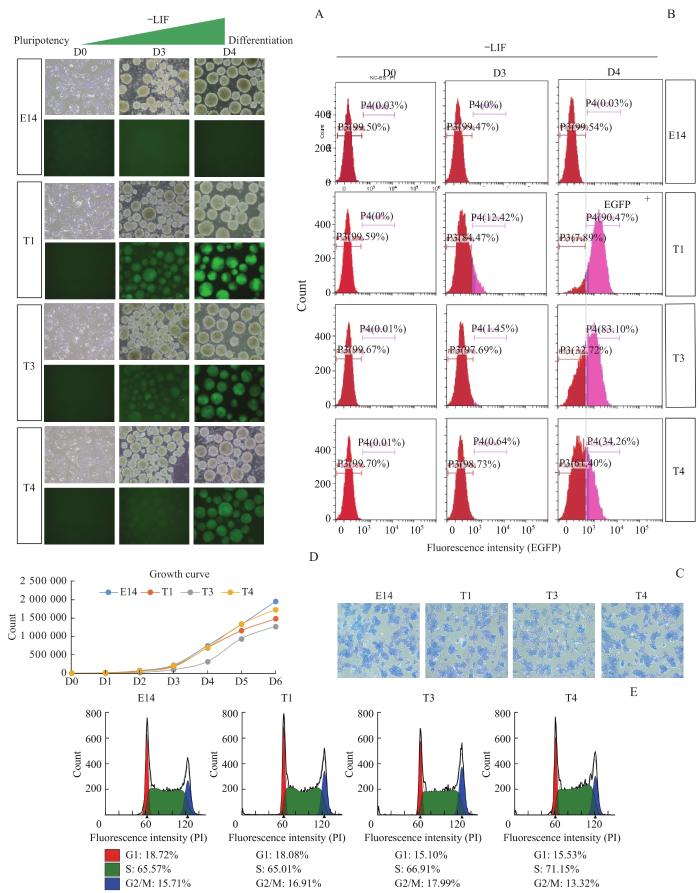
图5
mT-F2A-EGFP荧光报告干细胞系的功能检测
Note:A. Cell morphology and EGFP expression of E14, T1, T3 and T4 clones on Day 0 (D0), Day 3 (D3) and Day 4 (D4) of EB differentiation upon LIF removal (-LIF). All the microscopic pictures were taken under ×10 magnification. B. Flow cytometry analysis of E14, T1, T3 and T4 on D0, D3 and D4 of EB differentiation upon LIF removal (-LIF). Pink indicates EGFP+ cells, and red indicates EGFP- cells. C. AP staining of E14, T1, T3, and T4 cells. All the microscopic pictures were taken under ×10 magnification. D. The growth curve of E14, T1, T3 and T4. E. Cell cycle analysis of E14, T1, T3 and T4 cells.
Fig 5
Function test of mT-F2A-EGFP clones
2.6 应用mT-F2A-EGFP报告系统分选中内胚层细胞
在去除LIF的4 d中,EB细胞可以向外胚层和中内胚层分化,其中还包含了少量未分化的干细胞和2CLC细胞。因此,如果想从多谱系的EB中分选和鉴定T基因高表达的中内胚层细胞群,可以利用mT-F2A-EGFP报告基因的荧光表达特性来实现。本研究中T1克隆已被证实是最能反映T基因且不影响干细胞特性的报告细胞系。以分化4 d的E14细胞作为阴性对照,对分化D4的T1细胞系进行流式细胞分选,分别收集EGFP+和EGFP-细胞群(图6A)。然后,分别提取EGFP+和EGFP- 2组细胞的RNA,并进行了RT-qPCR检测(图6B)。结果显示,EGFP+细胞主要高表达中内胚层标志基因T、Eomes和Gsc,而EGFP-细胞则表达多种谱系标志物,例如多能性标志物Sox2,外胚层标志物配对盒因子6(paired box 6,Pax6)和Sox1以及2CLC标志基因Dux、Zscan4d和Zscan4(图6B)。这说明本研究构建的mT-F2A-EGFP报告系统T1克隆能正确反映内源性T基因的表达变化,在EB分化中特异性标记中内胚层细胞。
图6
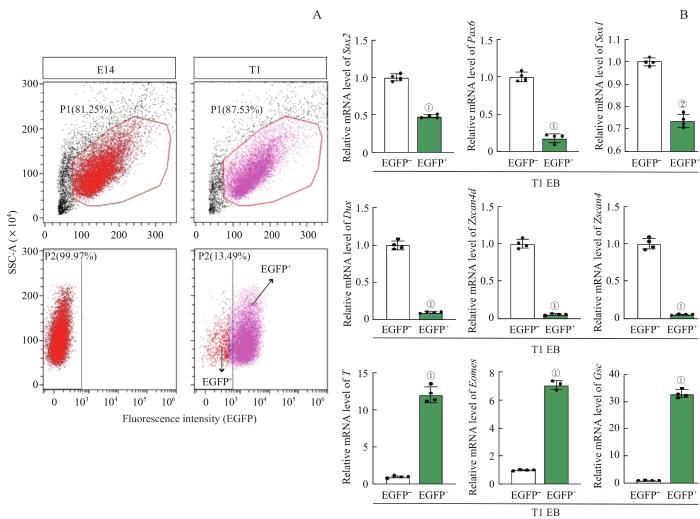
图6
应用mT-F2A-EGFP报告系统分选中内胚层细胞
Note:A. Flow cytometry analysis of E14 and T1 on EB differentiation day 4. Pink represents EGFP+ cells, and red represents EGFP- cells. B. Both EGFP+ and EGFP- cells of T1 on EB differentiation day 4 were collected by flow cytometry for RNA extraction. Relative mRNA levels of pluripotent marker Sox2, ectoderm markers Pax6 and Sox1, 2CLC markers Dux, Zscan4d and Zscan4, and mesendoderm markers T, Eomes and Gsc of EGFP+ and EGFP- cells were detected by RT-qPCR. ① P=0.000, ②P=0.005, compared with the EGFP- group.
Fig 6
mT-F2A-EGFP fluorescent reporter stem cell line used to sort mesendoderm cell populations
3 讨论
多能干细胞具有强大的自我更新和多向分化潜能,因此,在临床上有广泛的应用前景。然而,由于获取途径困难、培养条件高和分化异质性等问题,干细胞目前在临床上的实际应用仍具有不小的挑战[12]。只有深入阐明多能干细胞复杂的调控机制和功能,才能够进一步推动发育生物学进展和疾病模型构建,并最终促进干细胞替代疗法真正服务于临床。
2012年,CRISPR基因编辑技术首次问世。它的出现为定点基因编辑技术带来了新的契机。Cas9蛋白由sgRNA引导至特定的基因位点并诱导双链断裂。Cas9系统的优势在于构建简单且效率较高,但其高脱靶率带来了应用上的局限性。之后的Cas9n技术在Cas9的基础上进行了改进,仅能切割与sgRNA结合的DNA单链,并能最大限度地减少因错配导致的脱靶效应。Cas9n蛋白进行切割之后,如果存在外源性修复模板,供体基因则可以通过HDR途径被引入细胞中[13]。目前,CRISPR技术已广泛应用于农业、医疗等多个领域。利用CRISPR技术,研究人员可以在动物模型、细胞和分子层面进行发育与遗传生物学的探究;可以改良作物基因创造出更优良的优势品种、燃料、食物以及新型生物材料[14];可以为疾病诊疗提供新方案,例如针对新型冠状病毒(2019-nCoV)检测及治疗的研发[15];还在干细胞研究和修正人类缺陷基因以治疗遗传疾病等方面开辟了新的道路[16-17]等。
在小鼠胚胎的早期发育过程中,转化生长因子-β(transforming growth factor-β,TGF-β)和Wnt(wingless and int-1)信号通路在中内胚层的命运抉择中扮演了重要的角色。但是,早期胚胎向中内胚层分化的确切分子机制仍亟待研究。mESC能在体外重现体内的胚层分化,但是EB分化早期产生的多谱系细胞在一定程度上影响了对中内胚层细胞的研究。T基因作为中内胚层分化的重要调控因子,可以抑制神经外胚层的形成,并对于启动和诱导中内胚层的分化必不可少。缺失T基因的胚胎无法形成近轴中胚层[18],并且在mESC中,T基因和Eomes的表达均早于其他中内胚层基因[6]。因此,T基因能正确、及时地反映mESC向中内胚层分化的情况。
本研究正是基于CRISPR/Cas9n和HDR技术,在mESC E14中成功构建了mT-F2A-EGFP荧光报告细胞系T1。F2A肽介导的核糖体跳跃实现了Brachyury和EGFP的同时翻译与表达,并且F2A-EGFP和T基因的重组并没有影响E14细胞的干性维持和分化潜能。通过监测和分析T1报告细胞系的荧光信号变化,可以实时追踪分化前后T基因的表达情况,并分选出T基因高表达的中内胚层细胞。这个报告系统的建立为后续研究干细胞E14在退出多能性和诱导中内胚层分化过程中的作用奠定了坚实的基础。若与前沿的基因库或者化学小分子库的筛选技术相结合,则能更进一步地阐明mESC早期发育过程的命运决定机制。
作者贡献声明
王靖怡完成细胞培养、分子实验和文章撰写,王琼负责论文整体构思与文章修改。所有作者均阅读并同意了最终稿件的提交。
AUTHOR's CONTRIBUTIONS
WANG Jingyi performed the cell culture, molecular experiments, and manuscript draft. WANG Qiong designed and supervised the project and revise the manuscript. Both the authors have read the last version of paper and consented for submission.
利益冲突声明
所有作者声明不存在利益冲突。
COMPETING INTERESTS
Both authors disclose no relevant conflict of interests.
参考文献

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}